
![]()
NOTE: If you are on Colab, Run this cell to install the package
!pip install git+https://github.com/ahmedsalim3/GDAP.git
import gdap
from gdap.version import __version__
print(__version__)
# Set the project working directory
import sys
import os
import joblib
pwd = os.path.abspath('../')
sys.path.insert(0, pwd)
import os
# Import dataset and client modules
from gdap.datasets.string_database import PPIData
from gdap.datasets.open_targets import BigQueryClient, GraphQLClient
from gdap.datasets.open_targets.queries import DIRECT_SCORES, INDIRECT_SCORES
# Import graph creation and visualization
from gdap.graphs import BiGraph
# Import edge-related feature engineering functions
from gdap.edges.edge_utils import *
# Import embedding models
from gdap.embeddings import Node2Vec, ProNE, GGVec, EmbeddingGenerator
# Import model selection functions
from gdap.models.models_dict import sklearn_models
from gdap.models.model_evaluation import ModelEvaluation
# Import prediction functions
from gdap.edges.edge_predictions import *
# ---------------------------------------------------------------------------
# MAIN CONFIGURATION: (IDEALLY THIS SHOULD BE READ FROM A CONFIGURATION FILE)
# ---------------------------------------------------------------------------
# Set disease EFO-ID
disease_id = 'MONDO_0008903' # lung cancer
# disease_id = 'EFO_0000319' # cardiovascular
params = {'disease_id': disease_id}
# Path to BigQuery credentials (if using BigQuery data)
os.environ["GOOGLE_APPLICATION_CREDENTIALS"] = pwd + "/src/open_targets/stemaway-d5a18133ff83.json"
# Maximum number of protein-protein interactions (PPI)
max_ppi_interactions = 5000000
# Set ratio of negative to positive samples for classification
negative_to_positive_ratio = 10
# Define data source (can be direct, indirect, or global scores)
data_source = "GraphQL_global_scores" # Options: "BigQuery_indirect_scores", "GraphQL_global_scores", "BigQuery_direct_scores"
# Specify train/test split ratio for model validation
test_size = 0.2
# Specify whether to split edges for later prediction
split_edges = True
# Define embedding method (options include various node embedding algorithms)
embedding_mode = "Node2Vec" # Options: 'Node2Vec', 'ProNE', 'GGVec', 'simple_node_embedding'
# Set model selection for classification
model_name = "Logistic_Regression" # Options: 'Random_Forest', 'Gradient_Boosting', 'SVM', 'Logistic_Regression'
# Define output directory for models, edges, and embeddings
output_dir = pwd + '/results/'
if not os.path.exists(output_dir):
os.makedirs(output_dir)
# ---------------------
# DATA PROCESSING
# ---------------------
# 1. Load and process PPI data
ppi_data = PPIData(max_ppi_interactions=max_ppi_interactions)
ppi_df = ppi_data.process_ppi_data()
ppi_df.head()
Final PPI data shape: (5000000, 3)
| GeneName1 | GeneName2 | combined_score | |
|---|---|---|---|
| 0 | ESPL1 | PTTG1 | 0.999 |
| 1 | TP53 | MDM2 | 0.999 |
| 2 | PRPF3 | PRPF4 | 0.999 |
| 3 | CDKN1B | SKP2 | 0.999 |
| 4 | MLST8 | MTOR | 0.999 |
# 2. Fetch data based on the selected data source
if data_source == "GraphQL_global_scores":
# (i) Fetch global scores using GraphQL - NOTE: This score is approximately equal to the indirect scores from BigQuery
graphql_client = GraphQLClient()
ot_df = graphql_client.fetch_full_data(disease_id)
print(f'Final disease data shape: {ot_df.shape}')
elif data_source == "BigQuery_direct_scores":
# (ii) Fetch direct scores from BigQuery
bq_client = BigQueryClient()
ot_df = bq_client.execute_query(DIRECT_SCORES, params)
print(f'Final disease data shape: {ot_df.shape}')
elif data_source == "BigQuery_direct_scores":
# (iii) Fetch indirect scores from BigQuery
bq_client = BigQueryClient()
ot_df = bq_client.execute_query(INDIRECT_SCORES, params)
print(f'Final disease data shape: {ot_df.shape}')
ot_df.head()
Final disease data shape: (11121, 22)
| disease_name | symbol | globalScore | chembl | cancer_gene_census | intogen | eva | eva_somatic | uniprot_literature | clingen | ... | slapenrich | crispr | progeny | europepmc | genomics_england | impc | uniprot_variants | reactome | ot_genetics_portal | gene_burden | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | lung cancer | EGFR | 0.906916 | 0.995822 | 0.944823 | 0.932142 | 0.914326 | 0.905564 | 0.827461 | 0.607931 | ... | 0.977654 | 0.411539 | 0.683457 | 0.999188 | NaN | NaN | NaN | NaN | NaN | NaN |
| 1 | lung cancer | KRAS | 0.846207 | 0.888429 | 0.922212 | 0.932901 | 0.746974 | 0.899640 | NaN | NaN | ... | 0.989270 | 0.427663 | NaN | 0.977495 | 0.607931 | 0.787009 | NaN | NaN | NaN | NaN |
| 2 | lung cancer | ERBB2 | 0.836995 | 0.977565 | 0.697803 | 0.501595 | NaN | 0.869666 | NaN | NaN | ... | 0.959996 | NaN | NaN | 0.987588 | NaN | NaN | 0.827461 | NaN | NaN | NaN |
| 3 | lung cancer | BRAF | 0.835626 | 0.593257 | 0.713548 | 0.870006 | 0.848561 | 0.903643 | 0.827461 | NaN | ... | 0.963010 | NaN | NaN | 0.978271 | 0.827461 | NaN | 0.906661 | NaN | NaN | NaN |
| 4 | lung cancer | ALK | 0.829722 | 0.984832 | 0.956649 | 0.652385 | NaN | 0.674400 | NaN | NaN | ... | NaN | NaN | NaN | 0.997136 | NaN | NaN | NaN | NaN | NaN | NaN |
5 rows × 22 columns
# ---------------------
# GRAPH CREATION
# ---------------------
# Create a bipartite graph using the fetched disease data and PPI
G, positive_edges, negative_edges = BiGraph.create_graph(
ot_df,
ppi_df,
negative_to_positive_ratio=negative_to_positive_ratio,
output_dir=output_dir
)
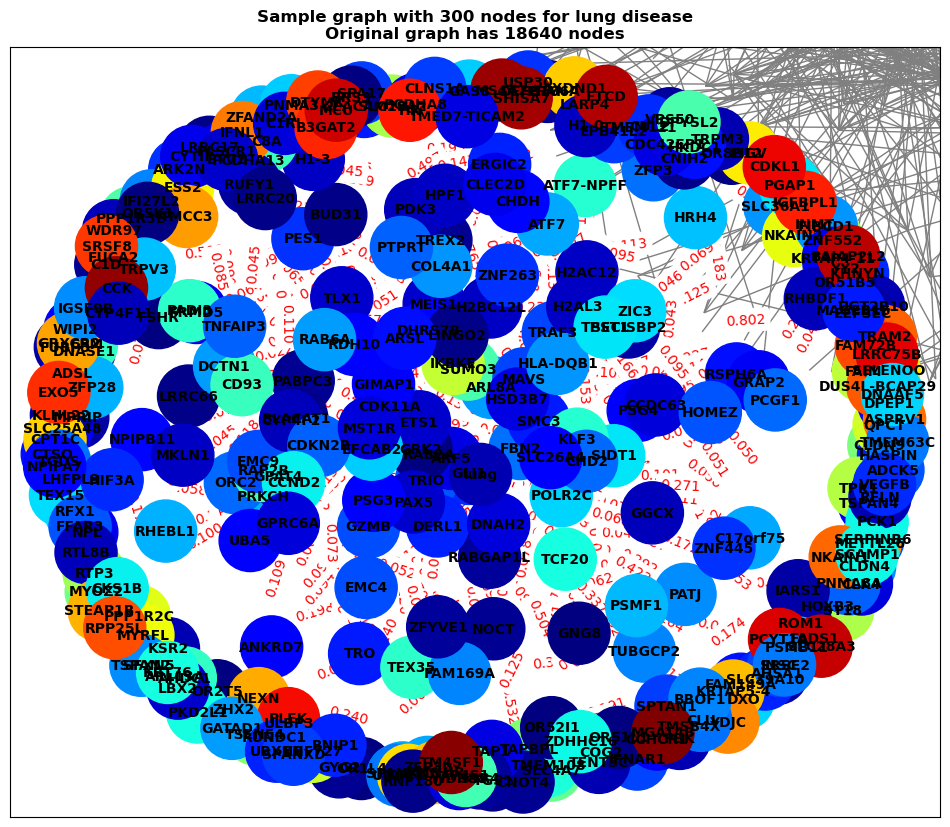
# Visualize a sample of the created graph
BiGraph.visualize_sample_graph(G, ot_df, node_size=300, figsize=(12,10), output_dir=output_dir)
Number of positive edges: 3271
Adding PPI edges: 100%|██████████| 5000000/5000000 [03:49<00:00, 21803.32it/s]
Number of negative edges: 32710
Sample graph saved to /home/ahmedsalim/projects/BI-ML_Disease-Prediction_2024/results/lung/network/lung_sample_graph_300_nodes.png
# ---------------------
# EMBEDDINGS
# ---------------------
if embedding_mode == "simple_node_embedding":
# Generate simple node embeddings
embeddings = EmbeddingGenerator.simple_node_embedding(G, dim=64)
disease_name = [d.split()[0] for d in ot_df['disease_name'].unique()][0]
print(f"\nEmbeddings for node '{disease_name}':\n{embeddings[str(disease_name)]}\n") # Show embeddings for disease node
elif embedding_mode == "Node2Vec":
# Train Node2Vec model on graph
n2v_model = Node2Vec(n_components=32, walklen=10)
print("Training Node2Vec model...")
embeddings = n2v_model.fit_transform(G)
node_to_index = map_nodes(G)
disease_name = [d.split()[0] for d in ot_df['disease_name'].unique()][0]
index = node_to_index[disease_name]
print(f"\nEmbeddings for node '{disease_name}':\n{embeddings[index]}")
# Save Node2Vec model and vectors
save_path = output_dir + disease_name + '/embedding_wheel/'
os.makedirs(save_path, exist_ok=True)
n2v_model.save(save_path + 'n2v_model')
n2v_model.save_vectors(save_path + "n2v_wheel_model.bin")
elif embedding_mode == "ProNE":
# Train ProNE model on graph
prone_model = ProNE(
n_components=32,
step=5,
mu=0.2,
theta=0.5,
exponent=0.75,
verbose=True
)
# Fit model to graph
print("Training ProNE model...")
embeddings = prone_model.fit_transform(G)
node_to_index = map_nodes(G)
disease_name = [d.split()[0] for d in ot_df['disease_name'].unique()][0]
index = node_to_index[disease_name]
print(f"Embeddings for node '{disease_name}':\n{embeddings[index]}")
# Save ProNE model and vectors
save_path = output_dir + disease_name + '/embedding_wheel/'
os.makedirs(save_path, exist_ok=True)
prone_model.save(save_path + 'prone_model')
ProNE.save_vectors(prone_model, save_path + "prone_wheel_model.bin")
print(f"Vectors/model saved to {save_path}")
elif embedding_mode == "GGVec":
# Train GGVec model on graph
ggvec_model = GGVec(
n_components=64,
order=3,
verbose=True
)
print("Training ProNE model...")
embeddings = ggvec_model.fit_transform(G)
node_to_index = map_nodes(G)
disease_name = [d.split()[0] for d in ot_df['disease_name'].unique()][0]
index = node_to_index[disease_name]
print(f"Embeddings for node '{disease_name}':\n{embeddings[index]}")
# Save GGVec model and vectors
save_path = output_dir + disease_name + '/embedding_wheel/'
os.makedirs(save_path, exist_ok=True)
ggvec_model.save(save_path + 'ggvec_model')
GGVec.save_vectors(ggvec_model, save_path + "ggvec_wheel_model.bin")
print(f"Vectors/model saved to {save_path}")
Training Node2Vec model...
Making walks... Done, T=3.59
Mapping Walk Names... Done, T=1.28
Training W2V... Done, T=209.38
Embeddings for node 'lung':
[-0.31097943 -4.1660657 1.5496684 -1.0685883 2.562408 -2.9975126
-2.5209763 -0.8388808 -3.6062336 -0.8412157 -4.6320105 -2.2024732
-4.355687 0.36631376 -4.021329 2.085732 0.76215327 6.9542704
-1.1137671 0.09638791 -6.113797 -4.667676 6.5994883 -0.04933656
6.3962965 -1.4965913 1.5549873 5.1001816 -0.55333275 -2.575983
0.9434795 -5.9954247 ]
# ------------------------------
# FEATURE EXTRACTION AND LABELING
# ------------------------------
# Extract features and labels from edges and embeddings
if embedding_mode == "simple_node_embedding" and not split_edges:
# Option 1: Directly get feature labels from edges and embeddings (using simple node embedding function)
X, y = features_labels(positive_edges, negative_edges, embeddings)
print(f'Sample from X: {X[0:1]}')
print(f'Sample from y: {y[0:1]}')
X_train, y_train, X_val, y_val, X_test, y_test = split_data(X, y, test_size=test_size)
elif embedding_mode == "simple_node_embedding" and split_edges:
# Option 2: Split edges for prediction, and get features/labels from split edges (using simple node embedding function)
X, y, edges = features_labels_edges(positive_edges, negative_edges, embeddings, scale_features=False)
print(f'Sample from X: {X[0:1]}')
print(f'Sample from y: {y[0:1]}')
X_train, y_train, X_val, y_val, X_test, y_test, edges_train, edges_val, edges_test = split_edge_data(X, y, edges, test_size=test_size)
elif embedding_mode != "simple_node_embedding" and not split_edges:
# Option 1: Get feature labels using node indexes for advanced embeddings algo (Node2Vec, ProNE, etc.)
X, y = features_labels_idx(positive_edges, negative_edges, embeddings, node_to_index)
print(f'Sample from X: {X[0:1]}')
print(f'Sample from y: {y[0:1]}')
X_train, y_train, X_val, y_val, X_test, y_test = split_data(X, y, test_size=test_size)
elif embedding_mode != "simple_node_embedding" and split_edges:
# Option 2: Get feature labels from split edges using node indexes for advanced embeddings algo (Node2Vec, ProNE, etc.)
X, y, edges = features_labels_edges_idx(positive_edges, negative_edges, embeddings, node_to_index, scale_features=False)
print(f'Sample from X: {X[0:1]}')
print(f'Sample from y: {y[0:1]}')
X_train, y_train, X_val, y_val, X_test, y_test, edges_train, edges_val, edges_test = split_edge_data(X, y, edges, test_size=test_size)
Sample from X: [[ 8.61236155e-01 -6.55511522e+00 -8.31855774e-01 5.21890558e-02
1.67569840e+00 -5.91884911e-01 -6.37902927e+00 -6.18832707e-01
5.47733158e-03 -8.73816609e-01 -7.25519061e-01 -1.76219487e+00
5.87105227e+00 -6.33723438e-01 2.37660551e+00 5.33728790e+00
1.84228227e-01 2.37836685e+01 7.33176947e-01 1.12808414e-01
1.06719494e+01 -7.08747530e+00 5.26128721e+00 1.10933542e-01
-1.32738390e+01 1.49221152e-01 -6.33619055e-02 1.44341218e+00
9.06589255e-02 -3.27536798e+00 6.15195453e-01 3.40170169e+00]]
Sample from y: [1]
# ------------------------------
# MODEL SELECTION AND PREDICTIONS
# ------------------------------
# OPTION 1: Fit a classifier model based on selected model_name from model_selection file
model_name = model_name
model, cv_scores= train_model(models[model_name], X_train, y_train, model_name=model_name)
test_results, val_results = validate_model(model, X_test, y_test, X_val, y_val, threshold=0.5)
print(f"\n{model_name} (Test Set)")
for metric, value in test_results.items():
print(f"{metric}: {value:.4f}")
print(f"\n{model_name} (Validation Set):")
for metric, value in val_results.items():
print(f"{metric}: {value:.4f}")
# Save Model
models_path = output_dir + disease_name + '/classifier_models/'
os.makedirs(models_path, exist_ok=True)
joblib.dump(model, models_path + f'{model_name}.pkl')
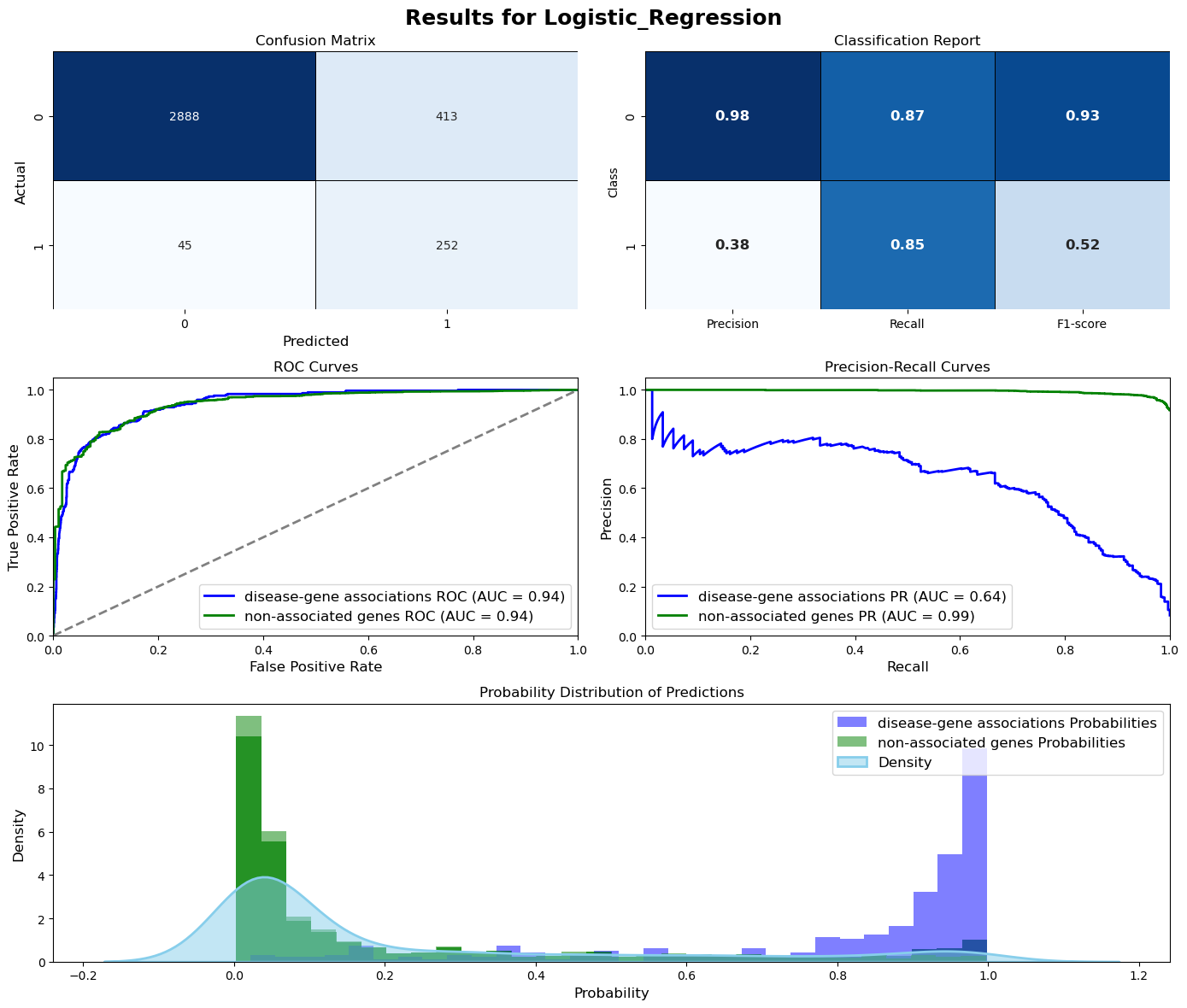
Logistic_Regression Cross-Validation Results:
Mean F1-score: 0.5531 (+/- 0.0375)
Logistic_Regression (Test Set)
Test Accuracy: 0.8697
Test Precision: 0.4274
Test Recall: 0.8595
Test F1-score: 0.5709
Test ROC-AUC: 0.9453
Test PR-AUC: 0.6856
Logistic_Regression (Validation Set):
Val Accuracy: 0.8727
Val Precision: 0.3789
Val Recall: 0.8485
Val F1-score: 0.5239
Val ROC-AUC: 0.9427
Val PR-AUC: 0.6416
['/home/ahmedsalim/projects/BI-ML_Disease-Prediction_2024/results/lung/classifier_models/Logistic_Regression.pkl']
Evaluation = ModelEvaluation(model, X_val, y_val, threshold=0.5, model_name=model_name, figsize=(14,12))
Evaluation.plot_evaluation()
# ----------------------------------
# PREDICTION RESULTS with Confidence
# -----------------------------------
threshold = 0.5
if split_edges:
associated_proteins, non_associated_proteins = predict(model, X_val, edges_val, threshold=threshold)
# Print results for associated proteins
print("\nAssociated Proteins:")
for i, (edge, (pred, confidence)) in enumerate(associated_proteins.items()):
if i < 5:
disease, protein = edge
print(f"Protein: {protein} (Disease: {disease}), Prediction: {pred}, Confidence Score: {confidence:.4f}")
# Print results for non-associated proteins
print("\nNon-Associated Proteins:")
for j, (edge, (pred, confidence)) in enumerate(non_associated_proteins.items()):
if j < 5:
disease, protein = edge
print(f"Protein: {protein} (Disease: {disease}), Prediction: {pred}, Confidence Score: {confidence:.4f}")
associated_df, non_associated_df = prediction_results(associated_proteins, non_associated_proteins, output_dir=output_dir)
print(f'\nProteins associated/non-associated to {disease_name} are saved to {output_dir}')
Associated Proteins:
Protein: TAS2R16 (Disease: lung), Prediction: 1, Confidence Score: 0.9775
Protein: CYP4F3 (Disease: lung), Prediction: 1, Confidence Score: 0.5712
Protein: HSD17B12 (Disease: lung), Prediction: 1, Confidence Score: 0.6226
Protein: CSN2 (Disease: lung), Prediction: 1, Confidence Score: 0.9423
Protein: NUP98 (Disease: lung), Prediction: 1, Confidence Score: 0.7276
Non-Associated Proteins:
Protein: WDR75 (Disease: lung), Prediction: 0, Confidence Score: 0.0714
Protein: RPL9 (Disease: lung), Prediction: 0, Confidence Score: 0.0078
Protein: COX5B (Disease: lung), Prediction: 0, Confidence Score: 0.3644
Protein: GEMIN8 (Disease: lung), Prediction: 0, Confidence Score: 0.3405
Protein: RPL26 (Disease: lung), Prediction: 0, Confidence Score: 0.0232
Proteins associated/non-associated to lung are saved to /home/ahmedsalim/projects/BI-ML_Disease-Prediction_2024/results/
associated_df.head()
| disease_name | symbol | prediction | confidence | |
|---|---|---|---|---|
| 313 | lung | NOTCH1 | 1 | 0.998299 |
| 31 | lung | MET | 1 | 0.997483 |
| 18 | lung | BMP2 | 1 | 0.997302 |
| 411 | lung | INHBB | 1 | 0.997241 |
| 153 | lung | SMAD3 | 1 | 0.996968 |
non_associated_df.head()
| disease_name | symbol | prediction | confidence | |
|---|---|---|---|---|
| 713 | lung | CNOT11 | 0 | 0.498954 |
| 336 | lung | FBXW4 | 0 | 0.497719 |
| 378 | lung | TMEM70 | 0 | 0.497098 |
| 610 | lung | DRAP1 | 0 | 0.496080 |
| 475 | lung | CXCL13 | 0 | 0.494857 |
# OPTION 2: Define your own model and train it, eg; gradient boosting model
gb_model = GradientBoostingClassifier(n_estimators=50, max_depth=3, learning_rate=0.1, random_state=42)
gb_name = "Gradient Boosting" # Optional
gb_model, gb_cv_scores= train_model(gb_model, X_train, y_train, model_name=gb_name)
gb_test_results, gb_val_results = validate_model(gb_model, X_test, y_test, X_val, y_val, threshold=0.5)
print(f"\n{gb_name} (Test Set):")
for metric, value in gb_test_results.items():
print(f"{metric}: {value:.4f}")
print(f"\n{gb_name} (Validation Set):")
for metric, value in gb_val_results.items():
print(f"{metric}: {value:.4f}")
# Save Model
joblib.dump(gb_model, models_path + 'gb_classifier.pkl')
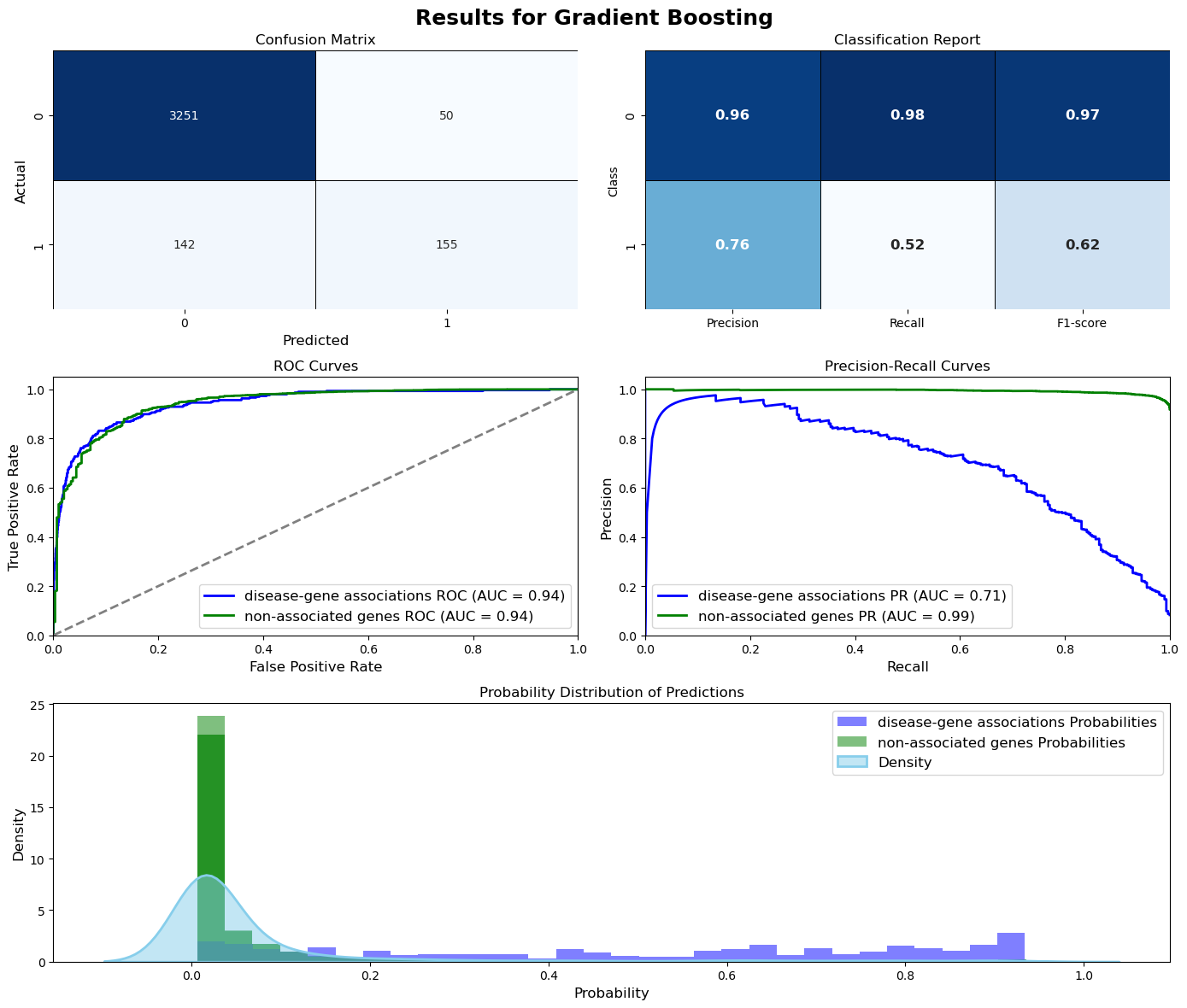
Gradient Boosting Cross-Validation Results:
Mean F1-score: 0.6578 (+/- 0.0306)
Gradient Boosting (Test Set):
Test Accuracy: 0.9433
Test Precision: 0.8630
Test Recall: 0.5207
Test F1-score: 0.6495
Test ROC-AUC: 0.9559
Test PR-AUC: 0.7871
Gradient Boosting (Validation Set):
Val Accuracy: 0.9466
Val Precision: 0.7561
Val Recall: 0.5219
Val F1-score: 0.6175
Val ROC-AUC: 0.9422
Val PR-AUC: 0.7103
['/home/ahmedsalim/projects/BI-ML_Disease-Prediction_2024/results/lung/classifier_models/gb_classifier.pkl']
Evaluation = ModelEvaluation(gb_model, X_val, y_val, threshold=0.5, model_name=gb_name, figsize=(14,12))
Evaluation.plot_evaluation()
threshold = 0.5
if split_edges:
associated_proteins, non_associated_proteins = predict(gb_model, X_val, edges_val, threshold=threshold)
associated_df, non_associated_df = prediction_results(associated_proteins, non_associated_proteins)
print(associated_df.head())
print()
print(non_associated_df.head())
disease_name symbol prediction confidence
27 lung AGT 1 0.935492
41 lung SLC17A6 1 0.934275
0 lung TAS2R16 1 0.930996
183 lung NDUFS1 1 0.930996
197 lung SNHG7 1 0.930996
disease_name symbol prediction confidence
820 lung GUCY1B1 0 0.497438
970 lung SRGAP3 0 0.487590
1012 lung CATSPER1 0 0.478803
1126 lung MGA 0 0.475078
347 lung NUTM2B 0 0.474420
# Example of simple dense model using sequential api
tf_model, history, acc, loss = train_tf_model(X_train, y_train, X_test, y_test, X_val, y_val, epochs=70)
print(f'Test Accuracy: {acc}\n Test Loss: {loss}')
# Save model
tf_model.save(models_path + 'tf.keras')
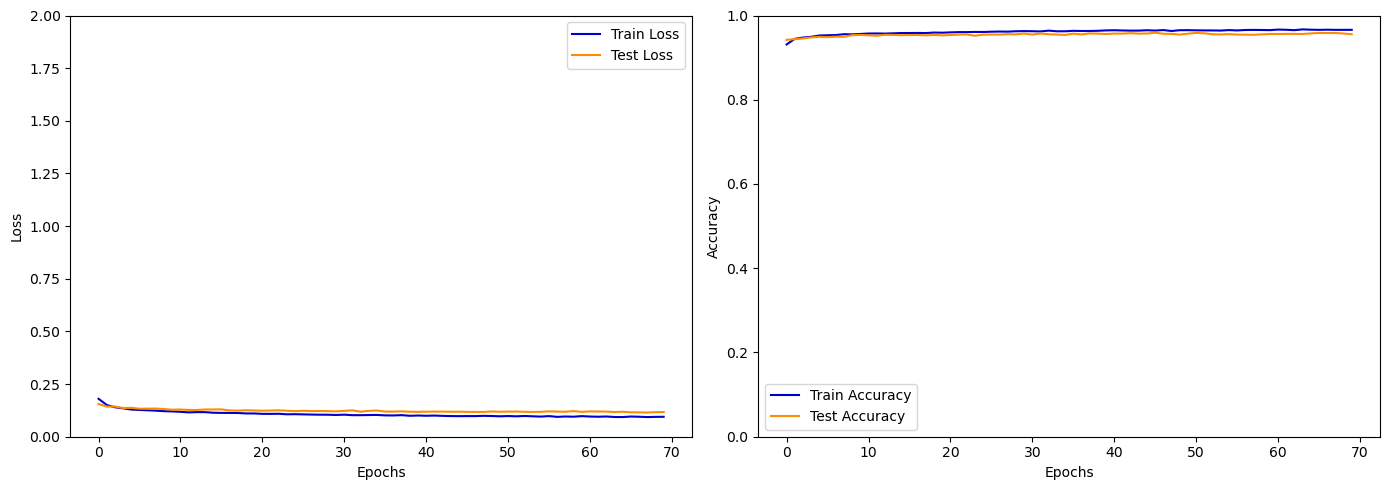
Training sequential API model...
[1m113/113[0m [32m━━━━━━━━━━━━━━━━━━━━[0m[37m[0m [1m1s[0m 5ms/step - accuracy: 0.9550 - loss: 0.1202
Test Accuracy: 0.9555, Test Loss: 0.1173
Test Accuracy: 0.9555432200431824
Test Loss: 0.11729290336370468
tf_test_results, tf_val_results = validate_model(model, X_test, y_test, X_val, y_val, threshold=0.5, is_tf_model=True)
print("Sequential Model (Test Set):")
for metric, value in gb_test_results.items():
print(f"{metric}: {value:.4f}")
print("\nSequential Model (Validation Set):")
for metric, value in gb_val_results.items():
print(f"{metric}: {value:.4f}")
Sequential Model (Test Set):
Test Accuracy: 0.9433
Test Precision: 0.8630
Test Recall: 0.5207
Test F1-score: 0.6495
Test ROC-AUC: 0.9559
Test PR-AUC: 0.7871
Sequential Model (Validation Set):
Val Accuracy: 0.9466
Val Precision: 0.7561
Val Recall: 0.5219
Val F1-score: 0.6175
Val ROC-AUC: 0.9422
Val PR-AUC: 0.7103
Evaluation = ModelEvaluation(tf_model, X_val, y_val, threshold=0.5, model_name="Sequential Model", figsize=(14,12),
is_tf_model=True, history=history, history_figsize=(14, 5))
Evaluation.plot_history()
Evaluation.plot_evaluation()
[1m113/113[0m [32m━━━━━━━━━━━━━━━━━━━━[0m[37m[0m [1m2s[0m 12ms/step
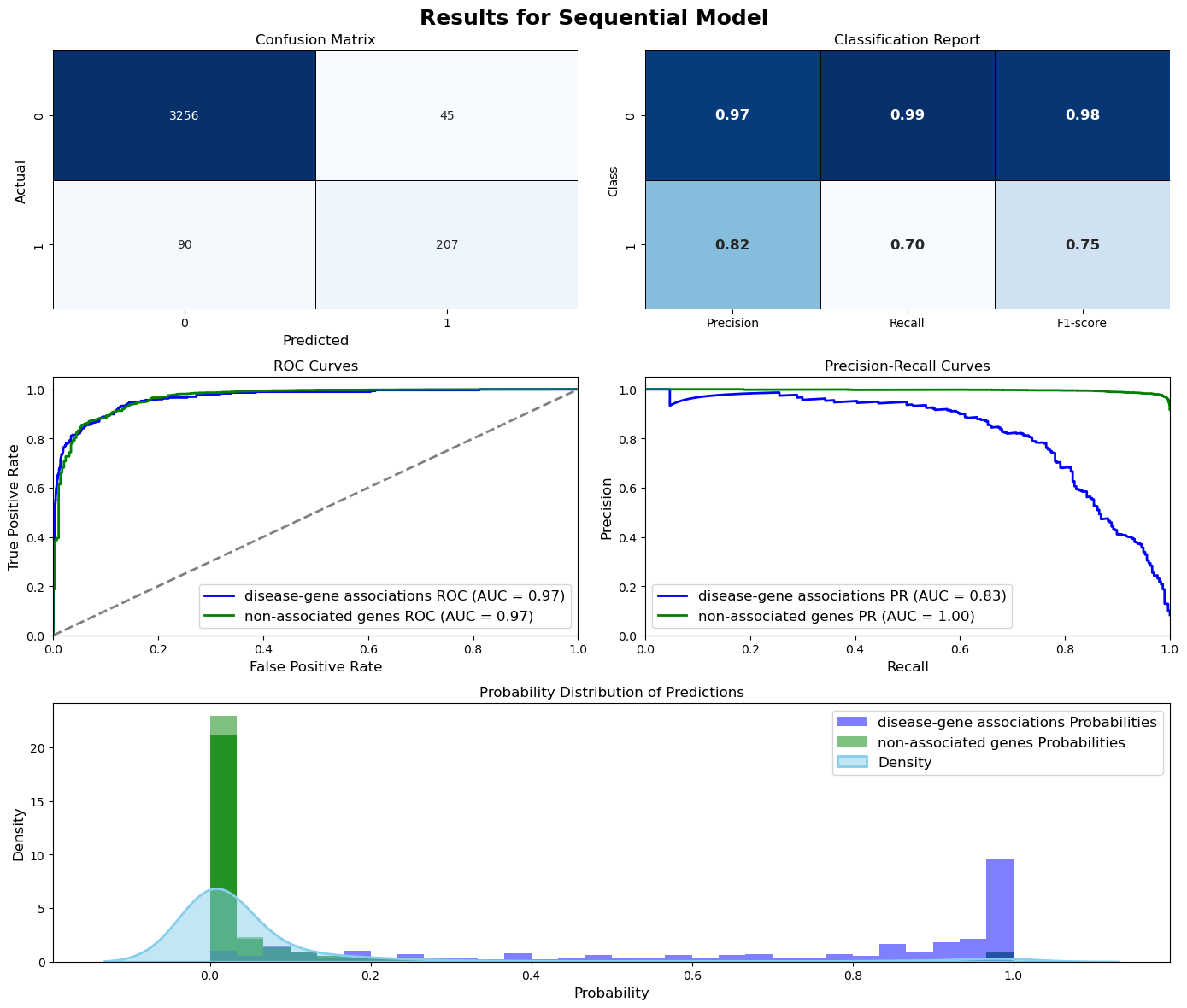
threshold = 0.5
if split_edges:
associated_proteins, non_associated_proteins = predict(tf_model, X_val, edges_val, threshold=threshold)
associated_df, non_associated_df = prediction_results(associated_proteins, non_associated_proteins)
associated_df
[1m113/113[0m [32m━━━━━━━━━━━━━━━━━━━━[0m[37m[0m [1m0s[0m 2ms/step
| disease_name | symbol | prediction | confidence | |
|---|---|---|---|---|
| 150 | lung | SYCE2 | 1 | 0.999978 |
| 56 | lung | FSHB | 1 | 0.999974 |
| 24 | lung | MYD88 | 1 | 0.999957 |
| 60 | lung | SLC17A6 | 1 | 0.999799 |
| 193 | lung | CATSPER1 | 1 | 0.999791 |
| ... | ... | ... | ... | ... |
| 113 | lung | CFB | 1 | 0.531065 |
| 73 | lung | CXCR2 | 1 | 0.525896 |
| 245 | lung | VCPKMT | 1 | 0.518479 |
| 68 | lung | RASSF1 | 1 | 0.514025 |
| 90 | lung | GIPC2 | 1 | 0.507189 |
248 rows × 4 columns
non_associated_df
| disease_name | symbol | prediction | confidence | |
|---|---|---|---|---|
| 529 | lung | CCR6 | 0 | 4.976931e-01 |
| 1022 | lung | EXO1 | 0 | 4.963719e-01 |
| 936 | lung | RRM2 | 0 | 4.926299e-01 |
| 519 | lung | NR0B2 | 0 | 4.912670e-01 |
| 933 | lung | XIAP | 0 | 4.745862e-01 |
| ... | ... | ... | ... | ... |
| 215 | lung | MRPL57 | 0 | 1.131001e-07 |
| 564 | lung | MRPS30 | 0 | 7.298207e-08 |
| 116 | lung | SF3B5 | 0 | 4.094883e-08 |
| 1081 | lung | MALSU1 | 0 | 1.030114e-09 |
| 28 | lung | NaN | 0 | 1.668466e-10 |
1090 rows × 4 columns