
Introduction
In this notebook, we explore the creation and analysis of disease-gene and protein-protein interaction networks using the NetworkX library. We will fetch relevant datasets, construct directed and undirected graphs, and visualize disease-specific networks, with a focus on breast cancer and other diseases. By the end, we will have a comprehensive understanding of network creation and analysis methods applicable to various diseases from open target platform.
![]()
Table of Contents
- Fetching Datasets
- Graph Creation and Visualization with NetworkX
- Disease-Specific Graph Construction
- Integrating Graphs and Class Definitions
We need to get the Open Targets disease data and then get the STRING Database for Protein-Protein Interaction (PPI)
We can either download the data manually, but we’ll use the GraphQL API and STRING API as they’re the fastest options to retrieve them
import requests
import json
from pprint import pprint
import pandas as pd
import networkx as nx
from networkx.algorithms import community
import matplotlib.pyplot as plt
Fetching Datasets
Open Target Dataset
Query Sourced from here and here
query = """
query DiseaseAssociationsQuery($efoId: String!){
disease(efoId: $efoId){
id
name
associatedTargets{
count
rows{
target{
id
approvedSymbol
}
score
datasourceScores {
id
score
}
}
}
}
}
"""
def fetch_graphql_data(disease_id):
"""
Fetch data from a GraphQL API endpoint.
"""
# Set variables
variables = {"efoId": disease_id}
# Set base URL of GraphQL API endpoint
base_url = "https://api.platform.opentargets.org/api/v4/graphql"
# Perform POST request and check status code of response
response = requests.post(base_url, json={"query": query, "variables": variables})
if response.status_code == 200:
print("request was successful\n")
# transform API response from JSON into Python dict
api_res = json.loads(response.text)
pprint(api_res)
return api_res
Let’s fetch the Breast Cancer dataset. The EFO ID for Breast Cancer is MONDO_0007254
disease_id = "MONDO_0007254" # you can find it at the top left of the open targets platform
api_res = fetch_graphql_data(disease_id)
request was successful
{'data': {'disease': {'associatedTargets': {'count': 11908,
'rows': [{'datasourceScores': [{'id': 'uniprot_variants',
'score': 0.9967809897717167},
{'id': 'gene_burden',
'score': 0.981427193451389},
{'id': 'genomics_england',
'score': 0.9779019396789099},
{'id': 'eva',
'score': 0.9699325219179143},
{'id': 'eva_somatic',
'score': 0.9488937677821665},
{'id': 'cancer_gene_census',
'score': 0.865017942479752},
{'id': 'uniprot_literature',
'score': 0.8274613634158176},
{'id': 'clingen',
'score': 0.607930797611621},
{'id': 'orphanet',
'score': 0.607930797611621},
{'id': 'slapenrich',
'score': 0.8979147935416834},
{'id': 'cancer_biomarkers',
'score': 0.865457038266544},
{'id': 'intogen',
'score': 0.336354528190285},
{'id': 'ot_genetics_portal',
'score': 0.3249228882691419},
{'id': 'europepmc',
'score': 0.9050938838031505},
{'id': 'impc',
'score': 0.4329809623969801}],
'score': 0.9246154877175718,
'target': {'approvedSymbol': 'BRCA2',
'id': 'ENSG00000139618'}},
{'datasourceScores': [{'id': 'uniprot_variants',
'score': 0.9967809897717167},
{'id': 'eva',
'score': 0.9699027100268988},
{'id': 'gene_burden',
'score': 0.9615415295186884},
{'id': 'eva_somatic',
'score': 0.9551136912636755},
{'id': 'genomics_england',
'score': 0.9471757543886921},
{'id': 'cancer_gene_census',
'score': 0.8655308214319849},
{'id': 'uniprot_literature',
'score': 0.8274613634158176},
{'id': 'orphanet',
'score': 0.607930797611621},
{'id': 'clingen',
'score': 0.607930797611621},
{'id': 'intogen',
'score': 0.5077719549344466},
{'id': 'slapenrich',
'score': 0.9626521671438224},
{'id': 'cancer_biomarkers',
'score': 0.865457038266544},
{'id': 'europepmc',
'score': 0.9967806662471856},
{'id': 'impc',
'score': 0.4942722213432534}],
'score': 0.9212307544907994,
'target': {'approvedSymbol': 'BRCA1',
'id': 'ENSG00000012048'}},
{'datasourceScores': [{'id': 'cancer_gene_census',
'score': 0.9632140851345072},
{'id': 'uniprot_variants',
'score': 0.9549947196932794},
{'id': 'intogen',
'score': 0.9509202575344607},
{'id': 'chembl',
'score': 0.9278946328772129},
{'id': 'eva_somatic',
'score': 0.8007968431539079},
{'id': 'eva',
'score': 0.7730988405360585},
{'id': 'slapenrich',
'score': 0.9865331671779851},
{'id': 'cancer_biomarkers',
'score': 0.9190679877429972},
{'id': 'crispr',
'score': 0.45050869819016554},
{'id': 'progeny',
'score': 0.5807359874260895},
{'id': 'europepmc',
'score': 0.988642821472146},
{'id': 'clingen',
'score': 0.006079307976116211}],
'score': 0.8793777133901898,
'target': {'approvedSymbol': 'PIK3CA',
'id': 'ENSG00000121879'}},
{'datasourceScores': [{'id': 'eva',
'score': 0.9685600208209743},
{'id': 'gene_burden',
'score': 0.9518332216356467},
{'id': 'genomics_england',
'score': 0.9190679877429972},
{'id': 'cancer_gene_census',
'score': 0.8412836902101531},
{'id': 'uniprot_literature',
'score': 0.8274613634158176},
{'id': 'clingen',
'score': 0.607930797611621},
{'id': 'slapenrich',
'score': 0.8493282943298344},
{'id': 'ot_genetics_portal',
'score': 0.29079990469976646},
{'id': 'europepmc',
'score': 0.8813600716365745}],
'score': 0.8672675131240247,
'target': {'approvedSymbol': 'PALB2',
'id': 'ENSG00000083093'}},
{'datasourceScores': [{'id': 'eva',
'score': 0.9493992387005677},
{'id': 'gene_burden',
'score': 0.9329896927619304},
{'id': 'uniprot_variants',
'score': 0.8897742701710087},
{'id': 'genomics_england',
'score': 0.8586178167934132},
{'id': 'uniprot_literature',
'score': 0.8274613634158176},
{'id': 'cancer_gene_census',
'score': 0.6893009908072479},
{'id': 'clingen',
'score': 0.607930797611621},
{'id': 'eva_somatic',
'score': 0.5015429080295873},
{'id': 'slapenrich',
'score': 0.9475493046633926},
{'id': 'ot_genetics_portal',
'score': 0.34792928780386473},
{'id': 'europepmc',
'score': 0.9684002733734489},
{'id': 'intogen',
'score': 0.15198269940290526},
{'id': 'chembl',
'score': 0.12158615952232422}],
'score': 0.8634848741187544,
'target': {'approvedSymbol': 'CHEK2',
'id': 'ENSG00000183765'}},
{'datasourceScores': [{'id': 'intogen',
'score': 0.9564265224205744},
{'id': 'cancer_gene_census',
'score': 0.9531985691504341},
{'id': 'eva',
'score': 0.9109719579436069},
{'id': 'eva_somatic',
'score': 0.8820907288419858},
{'id': 'genomics_england',
'score': 0.7599134970145264},
{'id': 'slapenrich',
'score': 0.9856315360164882},
{'id': 'progeny',
'score': 0.607930797611621},
{'id': 'cancer_biomarkers',
'score': 0.607930797611621},
{'id': 'europepmc',
'score': 0.9925763625311573},
{'id': 'impc',
'score': 0.7762948222349234},
{'id': 'chembl',
'score': 0.14353921610274387}],
'score': 0.8579753568583963,
'target': {'approvedSymbol': 'TP53',
'id': 'ENSG00000141510'}},
{'datasourceScores': [{'id': 'chembl',
'score': 0.9954471784803182},
{'id': 'cancer_gene_census',
'score': 0.9226214066290276},
{'id': 'ot_genetics_portal',
'score': 0.8077072080945117},
{'id': 'intogen',
'score': 0.7545563275197559},
{'id': 'cancer_biomarkers',
'score': 0.9607983220922475},
{'id': 'slapenrich',
'score': 0.8292004600368877},
{'id': 'crispr',
'score': 0.4028554954816608},
{'id': 'eva_somatic',
'score': 0.3039653988058105},
{'id': 'europepmc',
'score': 0.9975790652001129},
{'id': 'progeny',
'score': 0.3039653988058105},
{'id': 'impc',
'score': 0.2783107191466001},
{'id': 'eva',
'score': 0.015198269940290528}],
'score': 0.8579482544926643,
'target': {'approvedSymbol': 'ESR1',
'id': 'ENSG00000091831'}},
{'datasourceScores': [{'id': 'chembl',
'score': 0.9944690246399933},
{'id': 'cancer_gene_census',
'score': 0.9276008908162294},
{'id': 'intogen',
'score': 0.8005621897249015},
{'id': 'cancer_biomarkers',
'score': 0.9857024850308149},
{'id': 'slapenrich',
'score': 0.9742143913335374},
{'id': 'crispr',
'score': 0.39637088004277693},
{'id': 'europepmc',
'score': 0.9992089407387643},
{'id': 'eva_somatic',
'score': 0}],
'score': 0.8393681058533538,
'target': {'approvedSymbol': 'ERBB2',
'id': 'ENSG00000141736'}},
{'datasourceScores': [{'id': 'cancer_gene_census',
'score': 0.9199005839308011},
{'id': 'intogen',
'score': 0.9147281211319899},
{'id': 'reactome',
'score': 0.865457038266544},
{'id': 'uniprot_literature',
'score': 0.8274613634158176},
{'id': 'chembl',
'score': 0.6500589899523559},
{'id': 'eva_somatic',
'score': 0.547137717850459},
{'id': 'eva',
'score': 0.5073689115066987},
{'id': 'slapenrich',
'score': 0.9802615355154657},
{'id': 'crispr',
'score': 0.3275531137531414},
{'id': 'europepmc',
'score': 0.9827086927319217},
{'id': 'ot_genetics_portal',
'score': 0.07916000824617708}],
'score': 0.8282070633417251,
'target': {'approvedSymbol': 'AKT1',
'id': 'ENSG00000142208'}},
{'datasourceScores': [{'id': 'cancer_gene_census',
'score': 0.9470005150013617},
{'id': 'intogen',
'score': 0.9334985585431808},
{'id': 'eva',
'score': 0.9310298173424281},
{'id': 'eva_somatic',
'score': 0.6839221473130737},
{'id': 'slapenrich',
'score': 0.9390736181431255},
{'id': 'cancer_biomarkers',
'score': 0.607930797611621},
{'id': 'europepmc',
'score': 0.9705590991710802}],
'score': 0.8254207565099935,
'target': {'approvedSymbol': 'CDH1',
'id': 'ENSG00000039068'}},
{'datasourceScores': [{'id': 'eva',
'score': 0.9493909648924126},
{'id': 'genomics_england',
'score': 0.9066612367713315},
{'id': 'cancer_gene_census',
'score': 0.8920325429621649},
{'id': 'uniprot_variants',
'score': 0.8897742701710087},
{'id': 'slapenrich',
'score': 0.9326485111500797},
{'id': 'europepmc',
'score': 0.6886823067572628},
{'id': 'impc',
'score': 0.258005830506372},
{'id': 'clingen',
'score': 0.006079307976116211}],
'score': 0.8233869133963113,
'target': {'approvedSymbol': 'BRIP1',
'id': 'ENSG00000136492'}},
{'datasourceScores': [{'id': 'eva',
'score': 0.9687974527484059},
{'id': 'genomics_england',
'score': 0.7599134970145264},
{'id': 'cancer_gene_census',
'score': 0.7076059334305238},
{'id': 'clingen',
'score': 0.607930797611621},
{'id': 'eva_somatic',
'score': 0.57753425773104},
{'id': 'gene_burden',
'score': 0.5370693399711839},
{'id': 'slapenrich',
'score': 0.9721288826333171},
{'id': 'intogen',
'score': 0.3260714893140308},
{'id': 'europepmc',
'score': 0.9543985308894327},
{'id': 'impc',
'score': 0.6490645774656854},
{'id': 'ot_genetics_portal',
'score': 0.09417570446505588}],
'score': 0.8102877835482611,
'target': {'approvedSymbol': 'ATM',
'id': 'ENSG00000149311'}},
{'datasourceScores': [{'id': 'intogen',
'score': 0.9027520542982432},
{'id': 'cancer_gene_census',
'score': 0.866273997846718},
{'id': 'eva',
'score': 0.8570668490441947},
{'id': 'genomics_england',
'score': 0.607930797611621},
{'id': 'slapenrich',
'score': 0.9605410860851366},
{'id': 'cancer_biomarkers',
'score': 0.9190679877429972},
{'id': 'europepmc',
'score': 0.9829189382758448},
{'id': 'impc',
'score': 0.7483599311963033}],
'score': 0.784760888471985,
'target': {'approvedSymbol': 'PTEN',
'id': 'ENSG00000171862'}},
{'datasourceScores': [{'id': 'cancer_gene_census',
'score': 0.9315693279673053},
{'id': 'intogen',
'score': 0.9007870162776002},
{'id': 'ot_genetics_portal',
'score': 0.8006700483488376},
{'id': 'gene2phenotype',
'score': 0.3039653988058105},
{'id': 'europepmc',
'score': 0.38364098596622226}],
'score': 0.7707324681836835,
'target': {'approvedSymbol': 'MAP3K1',
'id': 'ENSG00000095015'}},
{'datasourceScores': [{'id': 'eva',
'score': 0.9462009100754374},
{'id': 'genomics_england',
'score': 0.8586178167934132},
{'id': 'orphanet',
'score': 0.607930797611621},
{'id': 'slapenrich',
'score': 0.9095618988667489},
{'id': 'europepmc',
'score': 0.8203383684006365},
{'id': 'clingen',
'score': 0.006079307976116211}],
'score': 0.7681561976356213,
'target': {'approvedSymbol': 'RAD51C',
'id': 'ENSG00000108384'}},
{'datasourceScores': [{'id': 'eva',
'score': 0.9489953381878606},
{'id': 'cancer_gene_census',
'score': 0.7032174930743333},
{'id': 'clingen',
'score': 0.607930797611621},
{'id': 'gene2phenotype',
'score': 0.607930797611621},
{'id': 'slapenrich',
'score': 0.9432831222921076},
{'id': 'gene_burden',
'score': 0.19577539648350917},
{'id': 'europepmc',
'score': 0.8922733459337661},
{'id': 'impc',
'score': 0.25824900282541663}],
'score': 0.7654432846523105,
'target': {'approvedSymbol': 'BARD1',
'id': 'ENSG00000138376'}},
{'datasourceScores': [{'id': 'chembl',
'score': 0.9863841154458036},
{'id': 'cancer_gene_census',
'score': 0.8699743018321302},
{'id': 'slapenrich',
'score': 0.8469682157917556},
{'id': 'europepmc',
'score': 0.3227423563847339}],
'score': 0.7629323355279769,
'target': {'approvedSymbol': 'CDK6',
'id': 'ENSG00000105810'}},
{'datasourceScores': [{'id': 'chembl',
'score': 0.9782080820264845},
{'id': 'cancer_gene_census',
'score': 0.7032174930743333},
{'id': 'slapenrich',
'score': 0.9776035037816935},
{'id': 'intogen',
'score': 0.35037268153398754},
{'id': 'crispr',
'score': 0.2942385060440246},
{'id': 'europepmc',
'score': 0.9922142106444732},
{'id': 'eva',
'score': 0.19453785523571876},
{'id': 'progeny',
'score': 0.3039653988058105},
{'id': 'eva_somatic',
'score': 0}],
'score': 0.7622532919844915,
'target': {'approvedSymbol': 'EGFR',
'id': 'ENSG00000146648'}},
{'datasourceScores': [{'id': 'eva',
'score': 0.9469210713496257},
{'id': 'genomics_england',
'score': 0.8274613634158176},
{'id': 'orphanet',
'score': 0.607930797611621},
{'id': 'slapenrich',
'score': 0.9089652610585701},
{'id': 'europepmc',
'score': 0.41246601446483044},
{'id': 'clingen',
'score': 0.006079307976116211}],
'score': 0.7618637638136027,
'target': {'approvedSymbol': 'RAD51D',
'id': 'ENSG00000185379'}},
{'datasourceScores': [{'id': 'chembl',
'score': 0.9103120255511722},
{'id': 'cancer_gene_census',
'score': 0.8611084598274582},
{'id': 'ot_genetics_portal',
'score': 0.6494039087995811},
{'id': 'slapenrich',
'score': 0.9748162620857227},
{'id': 'intogen',
'score': 0.3091165783782697},
{'id': 'cancer_biomarkers',
'score': 0.607930797611621},
{'id': 'europepmc',
'score': 0.7389422896398891}],
'score': 0.7611490409640307,
'target': {'approvedSymbol': 'ERBB4',
'id': 'ENSG00000178568'}},
{'datasourceScores': [{'id': 'chembl',
'score': 0.9769980215991244},
{'id': 'cancer_gene_census',
'score': 0.7001778390862753},
{'id': 'slapenrich',
'score': 0.8334267210607424},
{'id': 'intogen',
'score': 0.4115121385503686},
{'id': 'eva',
'score': 0.25963711147996316},
{'id': 'europepmc',
'score': 0.4497215895787084},
{'id': 'impc',
'score': 0.379166438470368}],
'score': 0.7529192716595609,
'target': {'approvedSymbol': 'POLD1',
'id': 'ENSG00000062822'}},
{'datasourceScores': [{'id': 'chembl',
'score': 0.9863841154458036},
{'id': 'cancer_gene_census',
'score': 0.6227068933869035},
{'id': 'slapenrich',
'score': 0.9229772476990096},
{'id': 'crispr',
'score': 0.3981682274459157},
{'id': 'europepmc',
'score': 0.9561795921185277}],
'score': 0.7452455272403391,
'target': {'approvedSymbol': 'CDK4',
'id': 'ENSG00000135446'}},
{'datasourceScores': [{'id': 'cancer_gene_census',
'score': 0.9226214066290276},
{'id': 'intogen',
'score': 0.8565451884252918},
{'id': 'slapenrich',
'score': 0.8472920286170607},
{'id': 'crispr',
'score': 0.3890757104714375},
{'id': 'cancer_biomarkers',
'score': 0.607930797611621},
{'id': 'europepmc',
'score': 0.9712342504116543},
{'id': 'eva_somatic',
'score': 0}],
'score': 0.7451414332495789,
'target': {'approvedSymbol': 'GATA3',
'id': 'ENSG00000107485'}},
{'datasourceScores': [{'id': 'cancer_gene_census',
'score': 0.9184184282751694},
{'id': 'intogen',
'score': 0.809326307468332},
{'id': 'ot_genetics_portal',
'score': 0.6891228181288519},
{'id': 'crispr',
'score': 0.29667022923447106},
{'id': 'europepmc',
'score': 0.777555491966681},
{'id': 'impc',
'score': 0.26779351634791904}],
'score': 0.7438454496469091,
'target': {'approvedSymbol': 'TBX3',
'id': 'ENSG00000135111'}},
{'datasourceScores': [{'id': 'chembl',
'score': 0.9769980215991244},
{'id': 'cancer_gene_census',
'score': 0.7050938226965915},
{'id': 'slapenrich',
'score': 0.8334267210607424},
{'id': 'eva',
'score': 0.2785700719367037},
{'id': 'europepmc',
'score': 0.19988561484069164}],
'score': 0.7408139382927514,
'target': {'approvedSymbol': 'POLE',
'id': 'ENSG00000177084'}}]},
'id': 'MONDO_0007254',
'name': 'breast cancer'}}}
# extract relevant data
associated_targets = api_res['data']['disease']['associatedTargets']['rows']
# normalize the JSON data
open_target_df = pd.json_normalize(
data=associated_targets,
record_path='datasourceScores', # the path to the records to flatten
meta=['target', 'score'], # the fields to extract from the parent record
record_prefix='datasourceScores_', # Prefix for fields from the records
errors='ignore'
)
# further normalize the nested target column
target_df = pd.json_normalize(open_target_df['target'])
open_target_df = open_target_df.drop(columns=['target'])
open_target_df = pd.concat([open_target_df, target_df], axis=1)
open_target_df.head()
| datasourceScores_id | datasourceScores_score | score | id | approvedSymbol | |
|---|---|---|---|---|---|
| 0 | uniprot_variants | 0.996781 | 0.924615 | ENSG00000139618 | BRCA2 |
| 1 | gene_burden | 0.981427 | 0.924615 | ENSG00000139618 | BRCA2 |
| 2 | genomics_england | 0.977902 | 0.924615 | ENSG00000139618 | BRCA2 |
| 3 | eva | 0.969933 | 0.924615 | ENSG00000139618 | BRCA2 |
| 4 | eva_somatic | 0.948894 | 0.924615 | ENSG00000139618 | BRCA2 |
agg_df = open_target_df.groupby('id').agg({
'approvedSymbol': 'first',
'score': 'mean'
}).reset_index()
agg_df.head()
| id | approvedSymbol | score | |
|---|---|---|---|
| 0 | ENSG00000012048 | BRCA1 | 0.921231 |
| 1 | ENSG00000039068 | CDH1 | 0.825421 |
| 2 | ENSG00000062822 | POLD1 | 0.752919 |
| 3 | ENSG00000083093 | PALB2 | 0.867268 |
| 4 | ENSG00000091831 | ESR1 | 0.857948 |
# Aggregate by ID
agg_df = open_target_df.groupby('id').agg({
'approvedSymbol': 'first',
'score': 'mean',
'datasourceScores_id': lambda x: list(x), # aggregate into a list
'datasourceScores_score': lambda x: list(x) # aggregate into a list
}).reset_index()
agg_df.head()
| id | approvedSymbol | score | datasourceScores_id | datasourceScores_score | |
|---|---|---|---|---|---|
| 0 | ENSG00000012048 | BRCA1 | 0.921231 | [uniprot_variants, eva, gene_burden, eva_somat... | [0.9967809897717167, 0.9699027100268988, 0.961... |
| 1 | ENSG00000039068 | CDH1 | 0.825421 | [cancer_gene_census, intogen, eva, eva_somatic... | [0.9470005150013617, 0.9334985585431808, 0.931... |
| 2 | ENSG00000062822 | POLD1 | 0.752919 | [chembl, cancer_gene_census, slapenrich, intog... | [0.9769980215991244, 0.7001778390862753, 0.833... |
| 3 | ENSG00000083093 | PALB2 | 0.867268 | [eva, gene_burden, genomics_england, cancer_ge... | [0.9685600208209743, 0.9518332216356467, 0.919... |
| 4 | ENSG00000091831 | ESR1 | 0.857948 | [chembl, cancer_gene_census, ot_genetics_porta... | [0.9954471784803182, 0.9226214066290276, 0.807... |
# These are the gene symbols associated with breast cancer disease
gene_symbols = agg_df['approvedSymbol'].tolist()
gene_symbols
['BRCA1',
'CDH1',
'POLD1',
'PALB2',
'ESR1',
'MAP3K1',
'CDK6',
'GATA3',
'RAD51C',
'PIK3CA',
'TBX3',
'CDK4',
'BRIP1',
'BARD1',
'BRCA2',
'TP53',
'ERBB2',
'AKT1',
'EGFR',
'ATM',
'PTEN',
'POLE',
'ERBB4',
'CHEK2',
'RAD51D']
# and these are the unique features associated with the gene symbols
features = []
for feature_list in agg_df['datasourceScores_id'].tolist():
for feature in feature_list:
if feature not in features:
features.append(feature)
features
['uniprot_variants',
'eva',
'gene_burden',
'eva_somatic',
'genomics_england',
'cancer_gene_census',
'uniprot_literature',
'orphanet',
'clingen',
'intogen',
'slapenrich',
'cancer_biomarkers',
'europepmc',
'impc',
'chembl',
'ot_genetics_portal',
'crispr',
'progeny',
'gene2phenotype',
'reactome']
def get_feature_score(df, gene_symbol, feature_key):
"""
Filters the DataFrame for a given gene symbol and
retrieves the score for the specified feature.
"""
gene_df = df[df['approvedSymbol'] == gene_symbol]
if gene_df.empty:
raise ValueError(f"No features found for the gene symbol: {gene_symbol}")
score = None
for _, row in gene_df.iterrows():
if feature_key in row['datasourceScores_id']:
if isinstance(row['datasourceScores_score'], float):
score = row['datasourceScores_score']
else:
index = row['datasourceScores_id'].index(feature_key)
score = row['datasourceScores_score'][index]
break
if score is None:
raise ValueError(f"Feature key '{feature_key}' not found for gene symbol: {gene_symbol}")
print(f"{feature_key} score for {gene_symbol}: {score}")
return score
# reference the (x1, y1) coordinates in the platform, as shown in the attached screenshot
score = get_feature_score(agg_df,"BRCA2", "ot_genetics_portal")
ot_genetics_portal score for BRCA2: 0.3249228882691419
STRING Database
source code used can be found here Getting String Network Interactions
https://string-db.org/api/[output-format]/network?identifiers=[your_identifiers]&[optional_parameters]
For the latter ML network, we can use this all partners of protein set
def fetch_string_data(my_genes, caller_identity="app.name"):
"""
Fetches data from the STRING API based on provided gene symbols
"""
string_api_url = "https://version-11-5.string-db.org/api"
output_format = "json"
method = "network"
## Construct URL
request_url = "/".join([string_api_url, output_format, method])
## Set parameters
params = {
"identifiers": "%0d".join(my_genes), # your protein
"species": 9606, # Human taxonomy, aka Homo sapiens
"caller_identity": caller_identity # your app name
}
## Call STRING
try:
res = requests.post(request_url, data=params)
res.raise_for_status()
data = res.json()
return data
except requests.exceptions.RequestException as e:
print(f"Request failed: {e}")
return None
except json.JSONDecodeError:
print("Failed to decode JSON response")
return None
# gene symbols are from our open targets
# print (gene_symbols)
my_genes = gene_symbols
data = fetch_string_data(my_genes)
STRING_df = pd.json_normalize(data, errors='ignore')
print(STRING_df.columns)
print(STRING_df.shape)
Index(['stringId_A', 'stringId_B', 'preferredName_A', 'preferredName_B',
'ncbiTaxonId', 'score', 'nscore', 'fscore', 'pscore', 'ascore',
'escore', 'dscore', 'tscore'],
dtype='object')
(396, 13)
| Field | Description |
|---|---|
| stringId_A | STRING identifier (protein A) |
| stringId_B | STRING identifier (protein B) |
| preferredName_A | common protein name (protein A) |
| preferredName_B | common protein name (protein B) |
| ncbiTaxonId | NCBI taxon identifier |
| score | combined score |
| nscore | gene neighborhood score |
| fscore | gene fusion score |
| pscore | phylogenetic profile score |
| ascore | coexpression score |
| escore | experimental score |
| dscore | database score |
| tscore | textmining score |
STRING_df.head()
| stringId_A | stringId_B | preferredName_A | preferredName_B | ncbiTaxonId | score | nscore | fscore | pscore | ascore | escore | dscore | tscore | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 9606.ENSP00000257566 | 9606.ENSP00000269305 | TBX3 | TP53 | 9606 | 0.400 | 0.0 | 0 | 0.0 | 0.000 | 0.000 | 0.0 | 0.400 |
| 1 | 9606.ENSP00000257566 | 9606.ENSP00000269305 | TBX3 | TP53 | 9606 | 0.400 | 0.0 | 0 | 0.0 | 0.000 | 0.000 | 0.0 | 0.400 |
| 2 | 9606.ENSP00000257566 | 9606.ENSP00000261769 | TBX3 | CDH1 | 9606 | 0.425 | 0.0 | 0 | 0.0 | 0.062 | 0.000 | 0.0 | 0.413 |
| 3 | 9606.ENSP00000257566 | 9606.ENSP00000261769 | TBX3 | CDH1 | 9606 | 0.425 | 0.0 | 0 | 0.0 | 0.062 | 0.000 | 0.0 | 0.413 |
| 4 | 9606.ENSP00000257566 | 9606.ENSP00000382423 | TBX3 | MAP3K1 | 9606 | 0.468 | 0.0 | 0 | 0.0 | 0.000 | 0.065 | 0.0 | 0.455 |
STRING_df.dtypes
stringId_A object
stringId_B object
preferredName_A object
preferredName_B object
ncbiTaxonId object
score float64
nscore float64
fscore int64
pscore float64
ascore float64
escore float64
dscore float64
tscore float64
dtype: object
df_filtered = STRING_df[STRING_df['escore'] > 0.4]
df_sorted = df_filtered.sort_values(by='escore', ascending=False)
df_sorted[['preferredName_A', 'preferredName_B', 'escore', 'score']].head()
| preferredName_A | preferredName_B | escore | score | |
|---|---|---|---|---|
| 91 | BARD1 | BRCA1 | 0.998 | 0.999 |
| 90 | BARD1 | BRCA1 | 0.998 | 0.999 |
| 252 | ERBB2 | EGFR | 0.982 | 0.998 |
| 253 | ERBB2 | EGFR | 0.982 | 0.998 |
| 120 | PALB2 | BRCA2 | 0.981 | 0.999 |
# fast overlap check for the BRCA1 gene
gene = 'BRCA1'
openTarget = open_target_df[open_target_df['approvedSymbol'] == gene]
openTarget.head()
| datasourceScores_id | datasourceScores_score | score | id | approvedSymbol | |
|---|---|---|---|---|---|
| 15 | uniprot_variants | 0.996781 | 0.921231 | ENSG00000012048 | BRCA1 |
| 16 | eva | 0.969903 | 0.921231 | ENSG00000012048 | BRCA1 |
| 17 | gene_burden | 0.961542 | 0.921231 | ENSG00000012048 | BRCA1 |
| 18 | eva_somatic | 0.955114 | 0.921231 | ENSG00000012048 | BRCA1 |
| 19 | genomics_england | 0.947176 | 0.921231 | ENSG00000012048 | BRCA1 |
StringDB = STRING_df[STRING_df['preferredName_A'] == gene]
StringDB.head()
| stringId_A | stringId_B | preferredName_A | preferredName_B | ncbiTaxonId | score | nscore | fscore | pscore | ascore | escore | dscore | tscore | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 392 | 9606.ENSP00000418960 | 9606.ENSP00000466399 | BRCA1 | RAD51D | 9606 | 0.825 | 0.0 | 0 | 0.0 | 0.126 | 0.095 | 0.0 | 0.797 |
| 393 | 9606.ENSP00000418960 | 9606.ENSP00000466399 | BRCA1 | RAD51D | 9606 | 0.825 | 0.0 | 0 | 0.0 | 0.126 | 0.095 | 0.0 | 0.797 |
| 394 | 9606.ENSP00000418960 | 9606.ENSP00000451828 | BRCA1 | AKT1 | 9606 | 0.987 | 0.0 | 0 | 0.0 | 0.000 | 0.835 | 0.8 | 0.657 |
| 395 | 9606.ENSP00000418960 | 9606.ENSP00000451828 | BRCA1 | AKT1 | 9606 | 0.987 | 0.0 | 0 | 0.0 | 0.000 | 0.835 | 0.8 | 0.657 |
StringDB2 = STRING_df[STRING_df['preferredName_B'] == gene]
StringDB2.head()
| stringId_A | stringId_B | preferredName_A | preferredName_B | ncbiTaxonId | score | nscore | fscore | pscore | ascore | escore | dscore | tscore | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 44 | 9606.ENSP00000257904 | 9606.ENSP00000418960 | CDK4 | BRCA1 | 9606 | 0.973 | 0.0 | 0 | 0.0 | 0.088 | 0.675 | 0.8 | 0.604 |
| 45 | 9606.ENSP00000257904 | 9606.ENSP00000418960 | CDK4 | BRCA1 | 9606 | 0.973 | 0.0 | 0 | 0.0 | 0.088 | 0.675 | 0.8 | 0.604 |
| 66 | 9606.ENSP00000259008 | 9606.ENSP00000418960 | BRIP1 | BRCA1 | 9606 | 0.999 | 0.0 | 0 | 0.0 | 0.212 | 0.981 | 0.8 | 0.989 |
| 67 | 9606.ENSP00000259008 | 9606.ENSP00000418960 | BRIP1 | BRCA1 | 9606 | 0.999 | 0.0 | 0 | 0.0 | 0.212 | 0.981 | 0.8 | 0.989 |
| 90 | 9606.ENSP00000260947 | 9606.ENSP00000418960 | BARD1 | BRCA1 | 9606 | 0.999 | 0.0 | 0 | 0.0 | 0.161 | 0.998 | 0.9 | 0.992 |
Graph Creation and Visualization with NetworkX
Creating Disease-Gene & Protein-Protein Interaction Network Graphs using Networkx
Tutorial: Directed and Undirected Graphs
in Networkx, there are two types of graphs, directed graphs have edges with a specific direction (from node u to node v), while undirected graphs treat edges as bidirectional
Creating Directed Graphs
def plot_DiGraph(
df: pd.DataFrame,
source_col: str,
target_col: str,
edge_attr_col: str,
figsize: tuple = (10, 8)
) -> None:
"""
Create and plot a directed graph from the given df
"""
# create a directed graph from the DataFrame
DiGraph = nx.from_pandas_edgelist(
df,
source=source_col,
target=target_col,
edge_attr=edge_attr_col,
create_using=nx.DiGraph() # notice the DiGraph class, aka DirectGraph
)
# plot the directed graph
plt.figure(figsize=figsize)
pos = nx.spring_layout(DiGraph)
nx.draw(
DiGraph,
pos,
with_labels=True,
node_color='lightblue',
edge_color='gray',
node_size=3000,
font_size=10,
font_weight='bold',
arrows=True
)
edge_labels = nx.get_edge_attributes(DiGraph, edge_attr_col)
nx.draw_networkx_edge_labels(DiGraph, pos, edge_labels=edge_labels)
plt.title(f'PPI Network ({DiGraph})')
plt.show()
return DiGraph
tut_STRING_df = STRING_df.copy()
# Select the first two gene symbols for the example
tut_gene = gene_symbols[0:2]
tut_gene
['BRCA1', 'CDH1']
# Filter the dataframe for interactions involving the selected genes
tut_STRING_df = tut_STRING_df[(tut_STRING_df['preferredName_A'].isin(tut_gene)) &
(tut_STRING_df['preferredName_B'].isin(gene_symbols))]
tut_STRING_df.head()
| stringId_A | stringId_B | preferredName_A | preferredName_B | ncbiTaxonId | score | nscore | fscore | pscore | ascore | escore | dscore | tscore | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 122 | 9606.ENSP00000261769 | 9606.ENSP00000278616 | CDH1 | ATM | 9606 | 0.459 | 0.0 | 0 | 0.0 | 0.0 | 0.000 | 0.0 | 0.459 |
| 123 | 9606.ENSP00000261769 | 9606.ENSP00000278616 | CDH1 | ATM | 9606 | 0.459 | 0.0 | 0 | 0.0 | 0.0 | 0.000 | 0.0 | 0.459 |
| 124 | 9606.ENSP00000261769 | 9606.ENSP00000369497 | CDH1 | BRCA2 | 9606 | 0.470 | 0.0 | 0 | 0.0 | 0.0 | 0.000 | 0.0 | 0.470 |
| 125 | 9606.ENSP00000261769 | 9606.ENSP00000369497 | CDH1 | BRCA2 | 9606 | 0.470 | 0.0 | 0 | 0.0 | 0.0 | 0.000 | 0.0 | 0.470 |
| 126 | 9606.ENSP00000261769 | 9606.ENSP00000342235 | CDH1 | ERBB4 | 9606 | 0.489 | 0.0 | 0 | 0.0 | 0.0 | 0.059 | 0.0 | 0.479 |
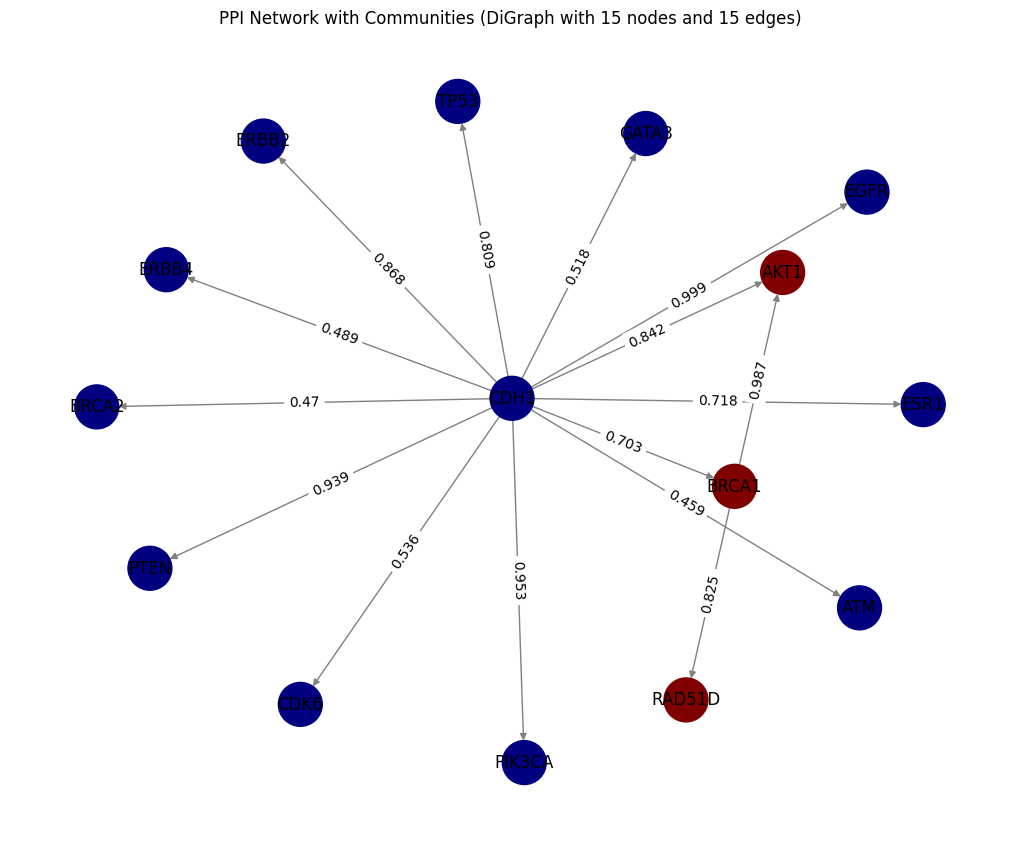
- In the STRING directed Graph the arrows indicate the direction of relationships from
preferredName_AtopreferredName_B. The weight represents as thescorevalue for each edge
Tut_STRING_DiGraph = plot_DiGraph(tut_STRING_df,'preferredName_A', 'preferredName_B', 'score')
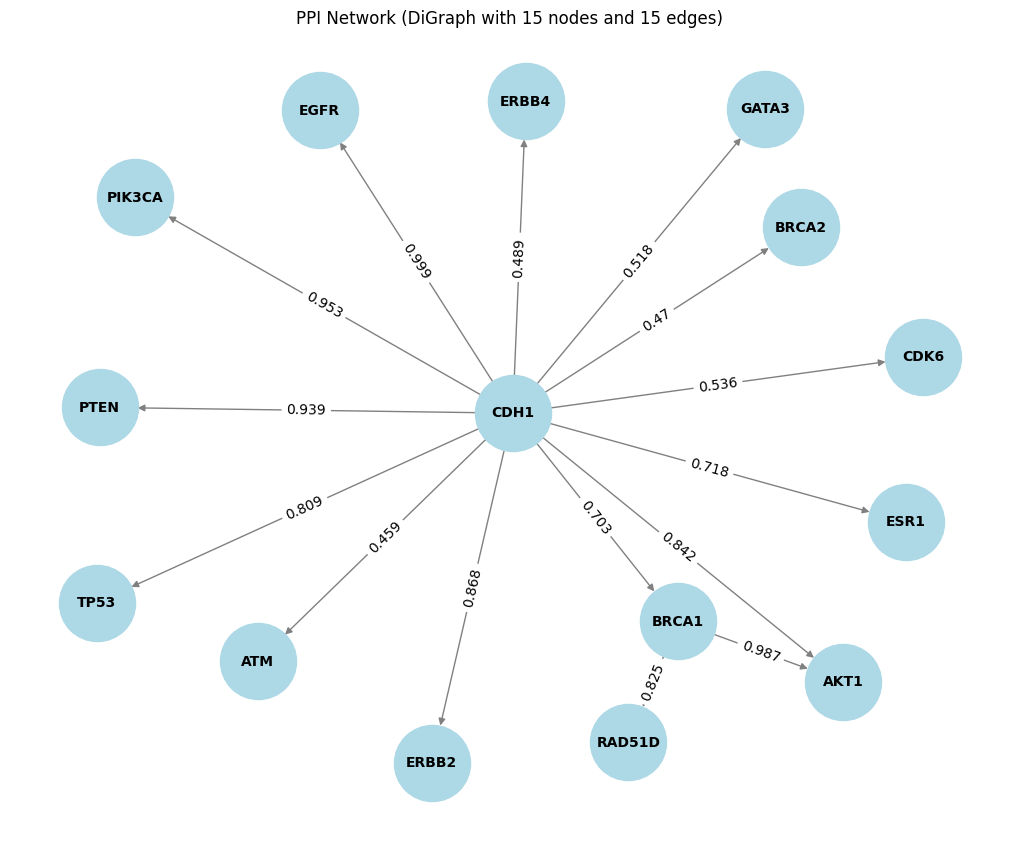
# similary we can see the directed graph in the open target dataset
tut_open_target_df = open_target_df.copy()
# Filter the df to include only rows with the a specific gene symbol
tut_open_target_df = tut_open_target_df[tut_open_target_df['approvedSymbol'] == 'BRCA1']
# Round the 'score' column to 2 decimal places
tut_open_target_df['datasourceScores_score'] = tut_open_target_df['datasourceScores_score'].round(2)
# plot the directed graph
Tut_open_target_DiGraph = plot_DiGraph(tut_open_target_df, 'datasourceScores_id', 'approvedSymbol', 'datasourceScores_score')
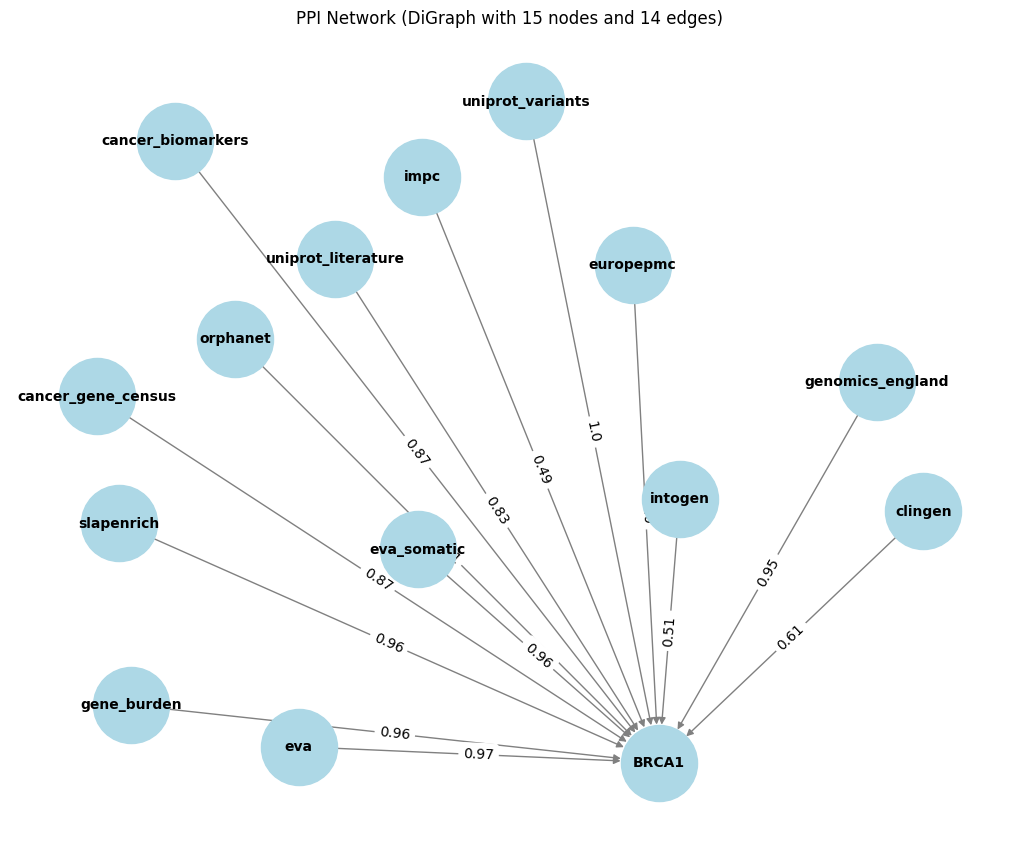
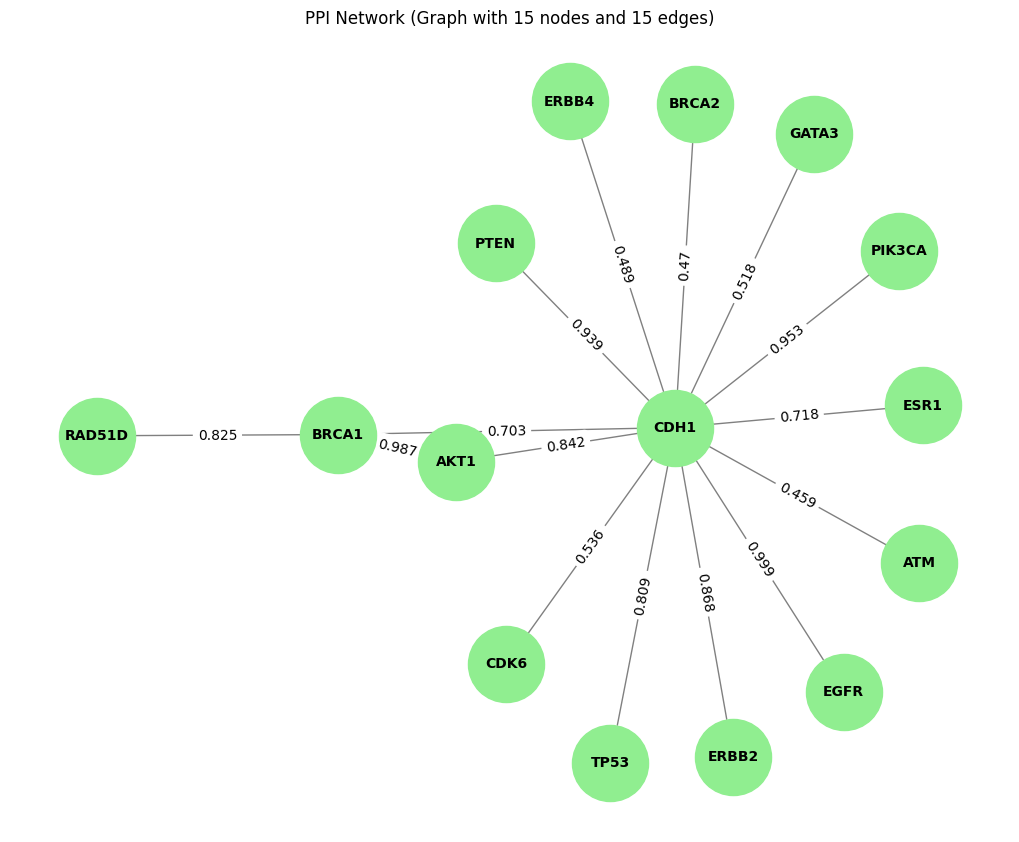
Creating Undirected Graphs
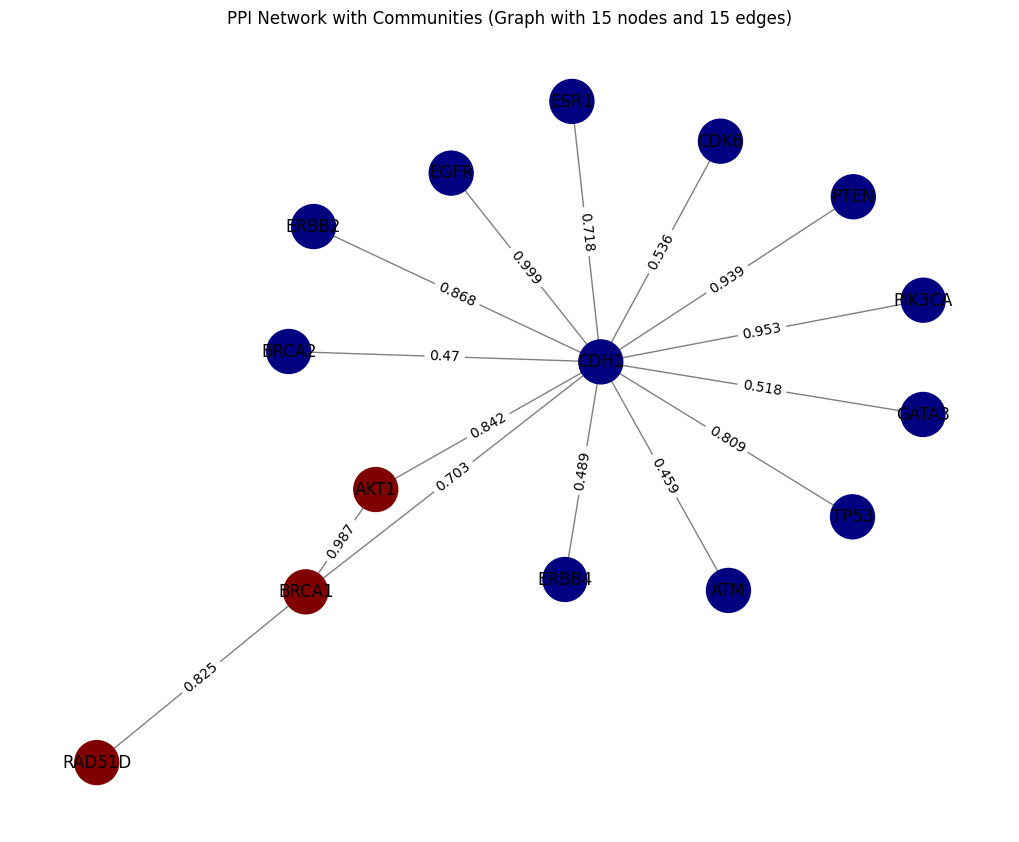
- In the undirected Graph, there are no arrows because edges are bidirectional. The weight (scores) remains unchanged, and edge direction is not considered
def plot_Graph(
df: pd.DataFrame,
source_col: str,
target_col: str,
edge_attr_col: str,
figsize: tuple = (10, 8)
) -> None:
"""
Create and plot a undirected graph from the given df
"""
# create a undirected graph from the DataFrame
G = nx.from_pandas_edgelist(
df,
source=source_col,
target=target_col,
edge_attr=edge_attr_col,
create_using=nx.Graph() # notice the undirect Graph
)
# plot the undirected graph
plt.figure(figsize=figsize)
pos = nx.spring_layout(G)
nx.draw(
G,
pos,
with_labels=True,
node_color='lightgreen',
edge_color='gray',
node_size=3000,
font_size=10,
font_weight='bold',
arrows=True
)
edge_labels = nx.get_edge_attributes(G, edge_attr_col)
nx.draw_networkx_edge_labels(G, pos, edge_labels=edge_labels)
plt.title(f'PPI Network ({G})')
plt.show()
return G
Tut_STRING_Graph = plot_Graph(tut_STRING_df,'preferredName_A', 'preferredName_B', 'score')
Tut_open_target_Graph = plot_Graph(tut_open_target_df, 'datasourceScores_id', 'approvedSymbol', 'datasourceScores_score')
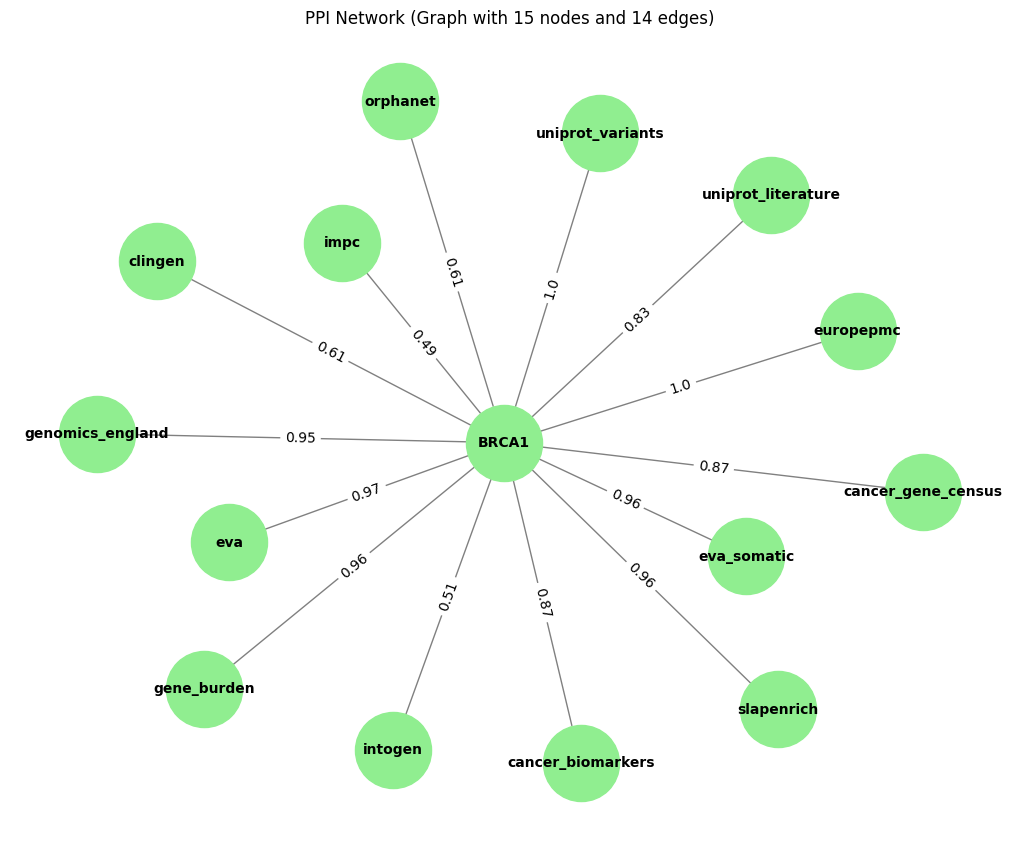
Which Graph we chose?
For a general PPI networks where interactions are typically mutual and direction is not that important, we use an undirected graph, as it’s the common choice for most PPI studies. However, if we ever need to direct these graphs, we now know where to look
Detecting Communities in Graphs
- Community detection identifies groups of nodes in a graph that are more connected to each other than to the rest of the network
- Each community is assigned a distinct color, so nodes in the same community (cluster) share the same color
- Colors help to easily visualize and distinguish different communities (clusters) within the network
def plot_community_detection(graph, edge_attr_col: str, figsize: tuple = (10, 8))-> None:
"""
plots graph with community detection
"""
communities = community.greedy_modularity_communities(graph)
# Color nodes by community
colors = [0] * graph.number_of_nodes()
for i, comm in enumerate(communities):
for node in comm:
colors[list(graph.nodes()).index(node)] = i
# Plotting
plt.figure(figsize=figsize)
pos = nx.spring_layout(graph)
nx.draw(
graph,
pos, with_labels=True,
node_color=colors,
cmap=plt.cm.jet, node_size=1000, edge_color='gray', arrows=True
)
edge_labels = nx.get_edge_attributes(graph, edge_attr_col)
nx.draw_networkx_edge_labels(graph, pos, edge_labels=edge_labels)
plt.title(f'PPI Network with Communities ({graph})')
plt.show()
plot_community_detection(Tut_STRING_DiGraph, edge_attr_col ='score')
plot_community_detection(Tut_STRING_Graph, edge_attr_col = 'score')
Disease-Specific Graph Construction
Building Breast Cancer Interaction Networks
we already fetched the breast cancer dataset in this section
# breast cancer dataset
open_target_df
| datasourceScores_id | datasourceScores_score | score | id | approvedSymbol | |
|---|---|---|---|---|---|
| 0 | uniprot_variants | 0.996781 | 0.924615 | ENSG00000139618 | BRCA2 |
| 1 | gene_burden | 0.981427 | 0.924615 | ENSG00000139618 | BRCA2 |
| 2 | genomics_england | 0.977902 | 0.924615 | ENSG00000139618 | BRCA2 |
| 3 | eva | 0.969933 | 0.924615 | ENSG00000139618 | BRCA2 |
| 4 | eva_somatic | 0.948894 | 0.924615 | ENSG00000139618 | BRCA2 |
| ... | ... | ... | ... | ... | ... |
| 209 | chembl | 0.976998 | 0.740814 | ENSG00000177084 | POLE |
| 210 | cancer_gene_census | 0.705094 | 0.740814 | ENSG00000177084 | POLE |
| 211 | slapenrich | 0.833427 | 0.740814 | ENSG00000177084 | POLE |
| 212 | eva | 0.278570 | 0.740814 | ENSG00000177084 | POLE |
| 213 | europepmc | 0.199886 | 0.740814 | ENSG00000177084 | POLE |
214 rows × 5 columns
open_target_df['datasourceScores_score'] = open_target_df['datasourceScores_score'].round(3)
STRING_df
| stringId_A | stringId_B | preferredName_A | preferredName_B | ncbiTaxonId | score | nscore | fscore | pscore | ascore | escore | dscore | tscore | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 9606.ENSP00000257566 | 9606.ENSP00000269305 | TBX3 | TP53 | 9606 | 0.400 | 0.0 | 0 | 0.0 | 0.000 | 0.000 | 0.0 | 0.400 |
| 1 | 9606.ENSP00000257566 | 9606.ENSP00000269305 | TBX3 | TP53 | 9606 | 0.400 | 0.0 | 0 | 0.0 | 0.000 | 0.000 | 0.0 | 0.400 |
| 2 | 9606.ENSP00000257566 | 9606.ENSP00000261769 | TBX3 | CDH1 | 9606 | 0.425 | 0.0 | 0 | 0.0 | 0.062 | 0.000 | 0.0 | 0.413 |
| 3 | 9606.ENSP00000257566 | 9606.ENSP00000261769 | TBX3 | CDH1 | 9606 | 0.425 | 0.0 | 0 | 0.0 | 0.062 | 0.000 | 0.0 | 0.413 |
| 4 | 9606.ENSP00000257566 | 9606.ENSP00000382423 | TBX3 | MAP3K1 | 9606 | 0.468 | 0.0 | 0 | 0.0 | 0.000 | 0.065 | 0.0 | 0.455 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 391 | 9606.ENSP00000406046 | 9606.ENSP00000418960 | POLD1 | BRCA1 | 9606 | 0.978 | 0.0 | 0 | 0.0 | 0.198 | 0.391 | 0.9 | 0.613 |
| 392 | 9606.ENSP00000418960 | 9606.ENSP00000466399 | BRCA1 | RAD51D | 9606 | 0.825 | 0.0 | 0 | 0.0 | 0.126 | 0.095 | 0.0 | 0.797 |
| 393 | 9606.ENSP00000418960 | 9606.ENSP00000466399 | BRCA1 | RAD51D | 9606 | 0.825 | 0.0 | 0 | 0.0 | 0.126 | 0.095 | 0.0 | 0.797 |
| 394 | 9606.ENSP00000418960 | 9606.ENSP00000451828 | BRCA1 | AKT1 | 9606 | 0.987 | 0.0 | 0 | 0.0 | 0.000 | 0.835 | 0.8 | 0.657 |
| 395 | 9606.ENSP00000418960 | 9606.ENSP00000451828 | BRCA1 | AKT1 | 9606 | 0.987 | 0.0 | 0 | 0.0 | 0.000 | 0.835 | 0.8 | 0.657 |
396 rows × 13 columns
Method 1: Using from_pandas_edgelist and compose
# create a graph for open target data
G = nx.from_pandas_edgelist(
open_target_df,
source='datasourceScores_id',
target='approvedSymbol',
edge_attr='datasourceScores_score',
# edge_attr=['datasourceScores_score', 'score'],
create_using=nx.Graph()
)
for u, v, data in G.edges(data=True):
data['weight'] = data.pop('datasourceScores_score') # Assign weight
data['type'] = 'disease-gene'
# Create a graph for the STRING dataframe
H = nx.from_pandas_edgelist(
STRING_df,
source='preferredName_A',
target='preferredName_B',
edge_attr='score',
# edge_attr=['score', 'nscore', 'fscore', 'pscore', 'ascore', 'escore', 'dscore', 'tscore'],
create_using=nx.Graph()
)
for u, v, data in H.edges(data=True):
data['weight'] = data.pop('score') # Assign weight
data['type'] = 'protein-protein'
# Merge the two graphs
G_composed = nx.compose(G, H)
plt.figure(figsize=(30, 20))
pos = nx.spring_layout(G_composed, seed=42, k=0.7, iterations=90) # Adjust 'k' and 'iterations' for better spacing
# Draw nodes with labels
nx.draw_networkx(G_composed, pos, with_labels=True, node_size=3000, font_size=10, font_weight='bold', edge_color='gray')
edge_labels = nx.get_edge_attributes(G_composed, 'weight')
nx.draw_networkx_edge_labels(G_composed, pos, edge_labels, font_color='red')
plt.axis('off')
plt.title(f"Breast Cancer Disease Associated Genes", fontsize=30, fontweight='bold')
plt.tight_layout()
# os.makedirs('assets', exist_ok=True)
# plt.savefig(f'assets/{disease_id}_{G_composed}.png')
plt.show()
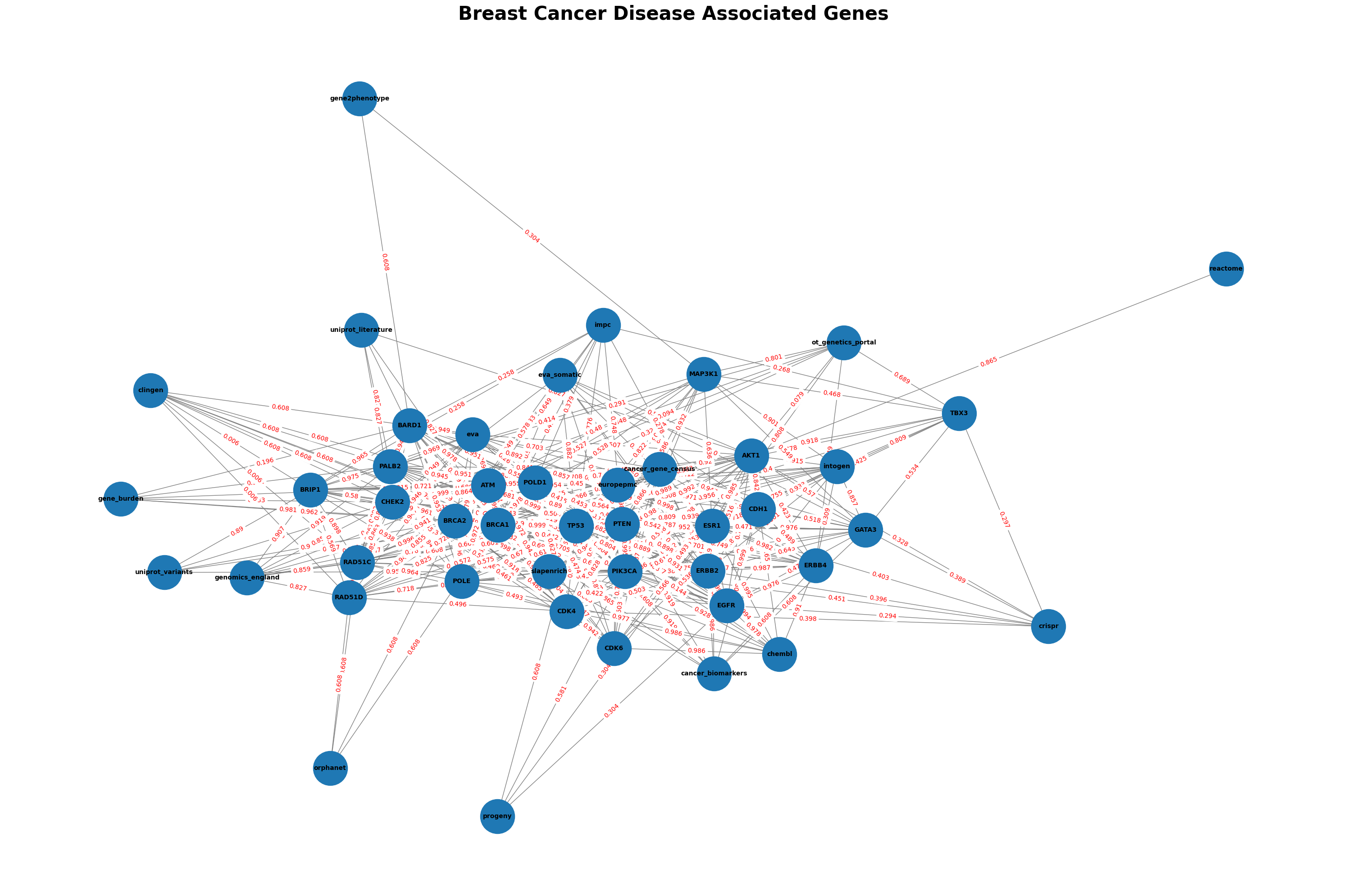
these edge attributes are preserved, and they can be accessed later using nx.get_edge_attributes with the specific attribute name score or datasourceScores_score
Method 2: Manually Adding Edges with add_edge and compose
G2 = nx.Graph()
for _, row in open_target_df.iterrows():
G2.add_edge(row['datasourceScores_id'],
row['approvedSymbol'],
weight=row['datasourceScores_score'],
type='disease-gene')
H2 = nx.Graph()
for _, row in STRING_df.iterrows():
H2.add_edge(row['preferredName_A'],
row['preferredName_B'],
weight=row['score'],
type='protein-protein')
# Merge the two graphs
G_composed2 = nx.compose(G2, H2)
plt.figure(figsize=(30, 20))
pos = nx.spring_layout(G_composed2, seed=42, k=0.9, iterations=100) # Adjust 'k' and 'iterations' for better spacing
# Draw nodes with labels
nx.draw_networkx(G_composed2, pos, with_labels=True, node_size=3000, font_size=10, font_weight='bold', edge_color='gray')
edge_labels = nx.get_edge_attributes(G_composed2, 'weight')
nx.draw_networkx_edge_labels(G_composed2, pos, edge_labels, font_color='red')
plt.axis('off')
plt.title(f"Breast Cancer Disease Associated Genes", fontsize=30, fontweight='bold')
plt.tight_layout()
# plt.savefig(f'assets/{disease_id}.png')
plt.show()
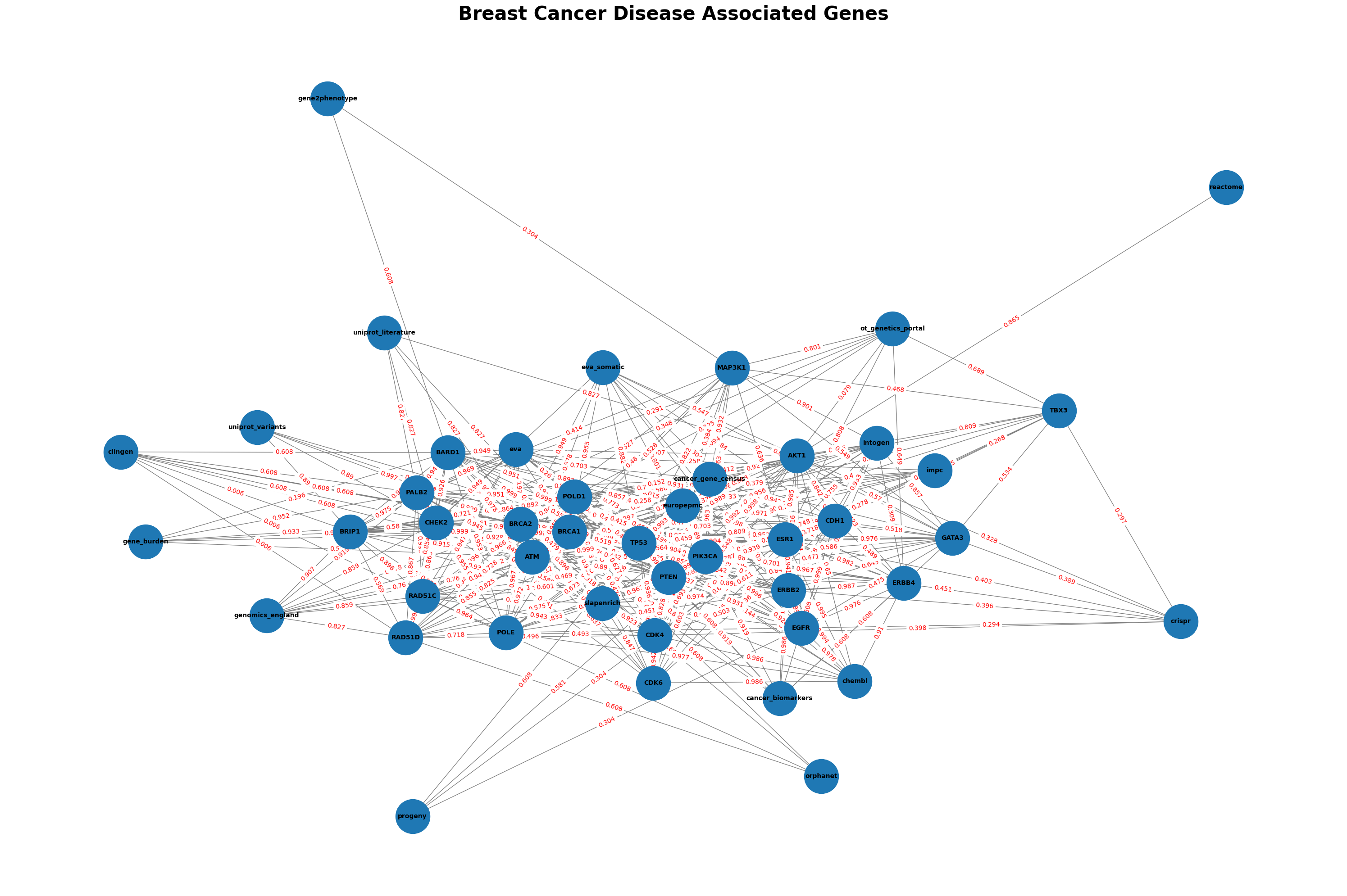
Comparing Node Connections Across Methods
def get_node_edges(graph, node, edge_type=None):
"""
Returns edges connected to specified nodes in a graph
optionally filtered by edge type
"""
edge_types = ['disease-gene', 'protein-protein']
if edge_type is not None and edge_type not in edge_types:
raise ValueError(f"Invalid edge type. Must be one of {edge_types}.")
edges = list(graph.edges(node, data=True))
if edge_type:
edges = [edge for edge in edges if edge[2].get('type') == edge_type]
return edges
def edge_to_tuple(edge):
"""Convert edge to a tuple with dictionaries as frozensets"""
u, v, attr = edge
attr_frozenset = frozenset(attr.items())
return (u, v, attr_frozenset)
node = 'ERBB4'
edges_composed = get_node_edges(G_composed, node, edge_type='protein-protein')
edges_composed2 = get_node_edges(G_composed2, node, edge_type='protein-protein')
print(f"Edges connected to {node} in {G_composed}:")
for edge in edges_composed[:5]:
print(edge)
print(f"\nEdges connected to {node} in {G_composed2}:")
for edge in edges_composed2[:5]:
print(edge)
edges_composed_set = set(edge_to_tuple(edge) for edge in edges_composed)
edges_composed2_set = set(edge_to_tuple(edge) for edge in edges_composed2)
if edges_composed_set == edges_composed2_set:
print("\nThe sets of edges are identical.")
Edges connected to ERBB4 in Graph with 45 nodes and 412 edges:
('ERBB4', 'TP53', {'weight': 0.701, 'type': 'protein-protein'})
('ERBB4', 'CDH1', {'weight': 0.489, 'type': 'protein-protein'})
('ERBB4', 'ESR1', {'weight': 0.982, 'type': 'protein-protein'})
('ERBB4', 'ERBB2', {'weight': 0.987, 'type': 'protein-protein'})
('ERBB4', 'AKT1', {'weight': 0.423, 'type': 'protein-protein'})
Edges connected to ERBB4 in Graph with 45 nodes and 412 edges:
('ERBB4', 'TP53', {'weight': 0.701, 'type': 'protein-protein'})
('ERBB4', 'CDH1', {'weight': 0.489, 'type': 'protein-protein'})
('ERBB4', 'ESR1', {'weight': 0.982, 'type': 'protein-protein'})
('ERBB4', 'ERBB2', {'weight': 0.987, 'type': 'protein-protein'})
('ERBB4', 'AKT1', {'weight': 0.423, 'type': 'protein-protein'})
The sets of edges are identical.
Integrating Graphs and Class Definitions
import sys
sys.path.append('../src')
from graph_composer import GraphComposer
Generating Graphs for Infectious Diseases
The dataset for the Infectious Disease is identified by EFO_0005741
disease_id = "EFO_0005741"
disease_name="Infectious"
Infectious = GraphComposer(disease_id, disease_name)
Infectious.process_all(plot=True)
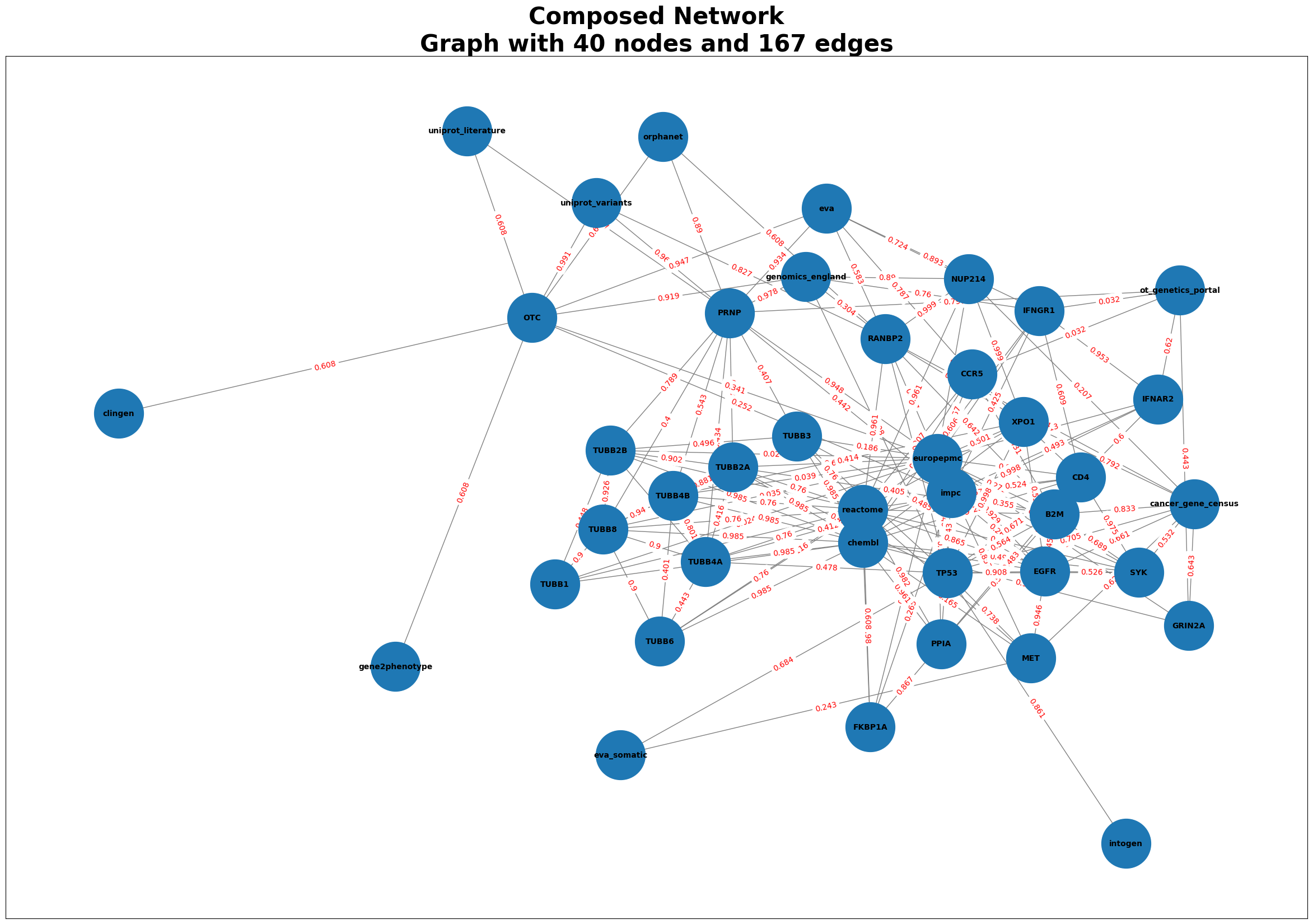
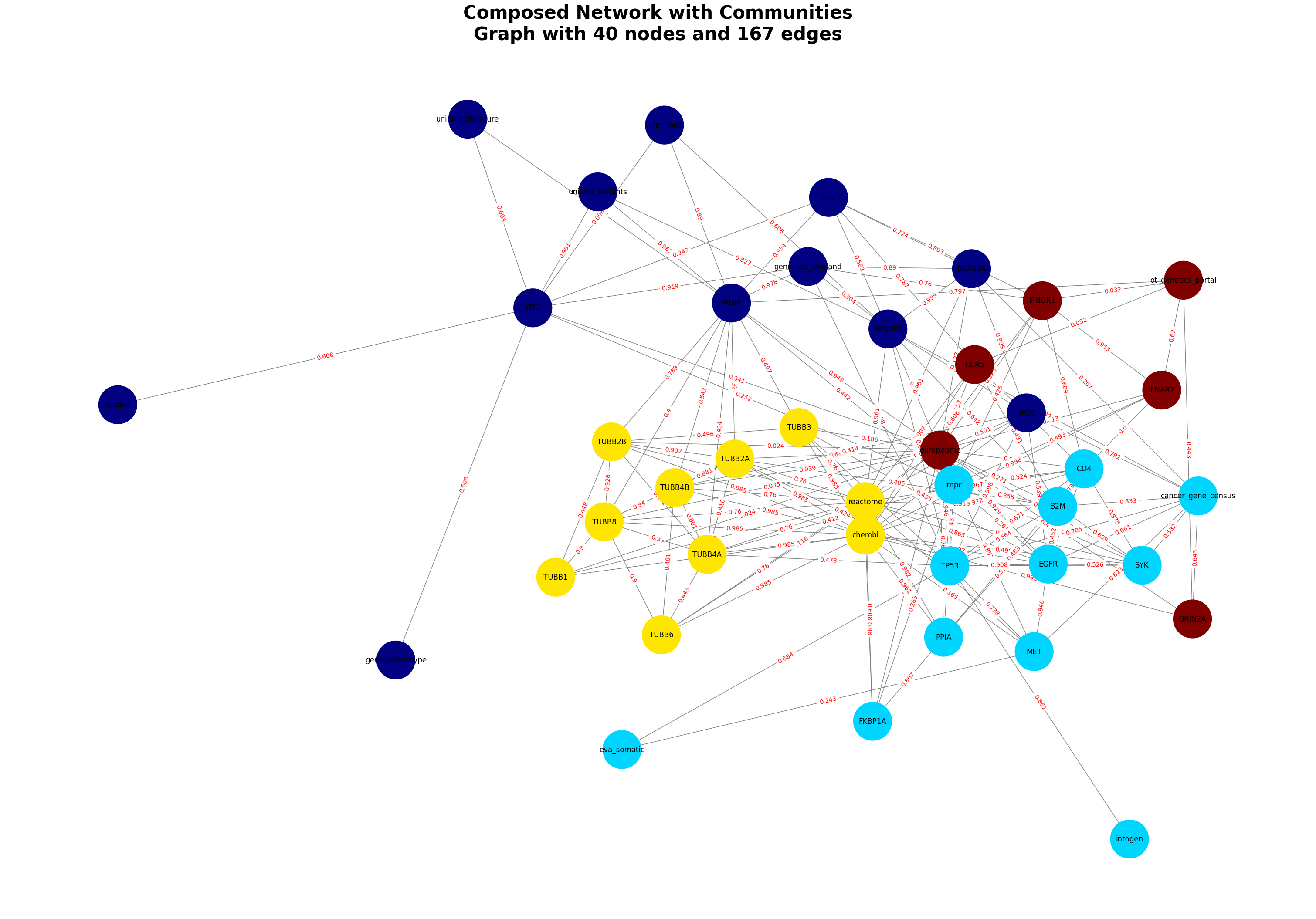
# Accessing the features, and genes
features = Infectious.features
df = Infectious.open_target_df
gene = Infectious.gene_symbols
score = get_feature_score(df, gene_symbol = 'PRNP', feature_key='orphanet')
orphanet score for PRNP: 0.89
merged_df = Infectious.get_merged_dataframe()
print(merged_df.describe)
print(merged_df.info())
<bound method NDFrame.describe of source target weight type
0 genomics_england PRNP 0.978 disease-gene
1 genomics_england OTC 0.919 disease-gene
2 genomics_england RANBP2 0.304 disease-gene
3 genomics_england NUP214 0.890 disease-gene
4 genomics_england TP53 0.608 disease-gene
.. ... ... ... ...
162 XPO1 EGFR 0.539 protein-protein
163 EGFR B2M 0.452 protein-protein
164 EGFR SYK 0.526 protein-protein
165 EGFR MET 0.946 protein-protein
166 SYK B2M 0.689 protein-protein
[167 rows x 4 columns]>
<class 'pandas.core.frame.DataFrame'>
RangeIndex: 167 entries, 0 to 166
Data columns (total 4 columns):
# Column Non-Null Count Dtype
--- ------ -------------- -----
0 source 167 non-null object
1 target 167 non-null object
2 weight 167 non-null float64
3 type 167 non-null object
dtypes: float64(1), object(3)
memory usage: 5.3+ KB
None
merged_df.head()
| source | target | weight | type | |
|---|---|---|---|---|
| 0 | genomics_england | PRNP | 0.978 | disease-gene |
| 1 | genomics_england | OTC | 0.919 | disease-gene |
| 2 | genomics_england | RANBP2 | 0.304 | disease-gene |
| 3 | genomics_england | NUP214 | 0.890 | disease-gene |
| 4 | genomics_england | TP53 | 0.608 | disease-gene |
Generating Graphs for Alzheimer’s Disease
The dataset for the Alzheimer Disease is identified by MONDO_0004975
disease_id = "MONDO_0004975"
Alzheimer = GraphComposer(disease_id, disease_name="Alzheimer")
Alzheimer.process_all(plot=True)
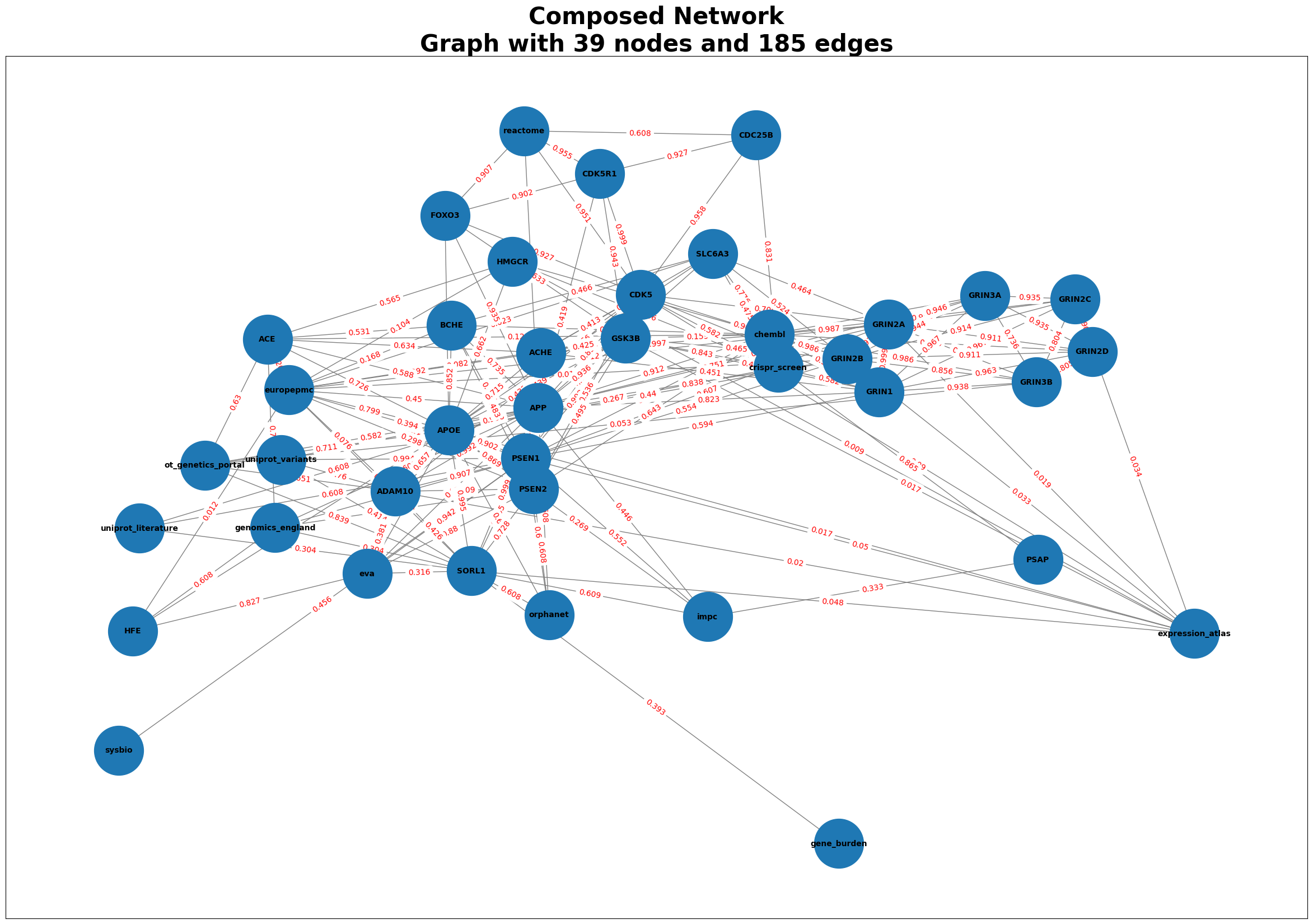
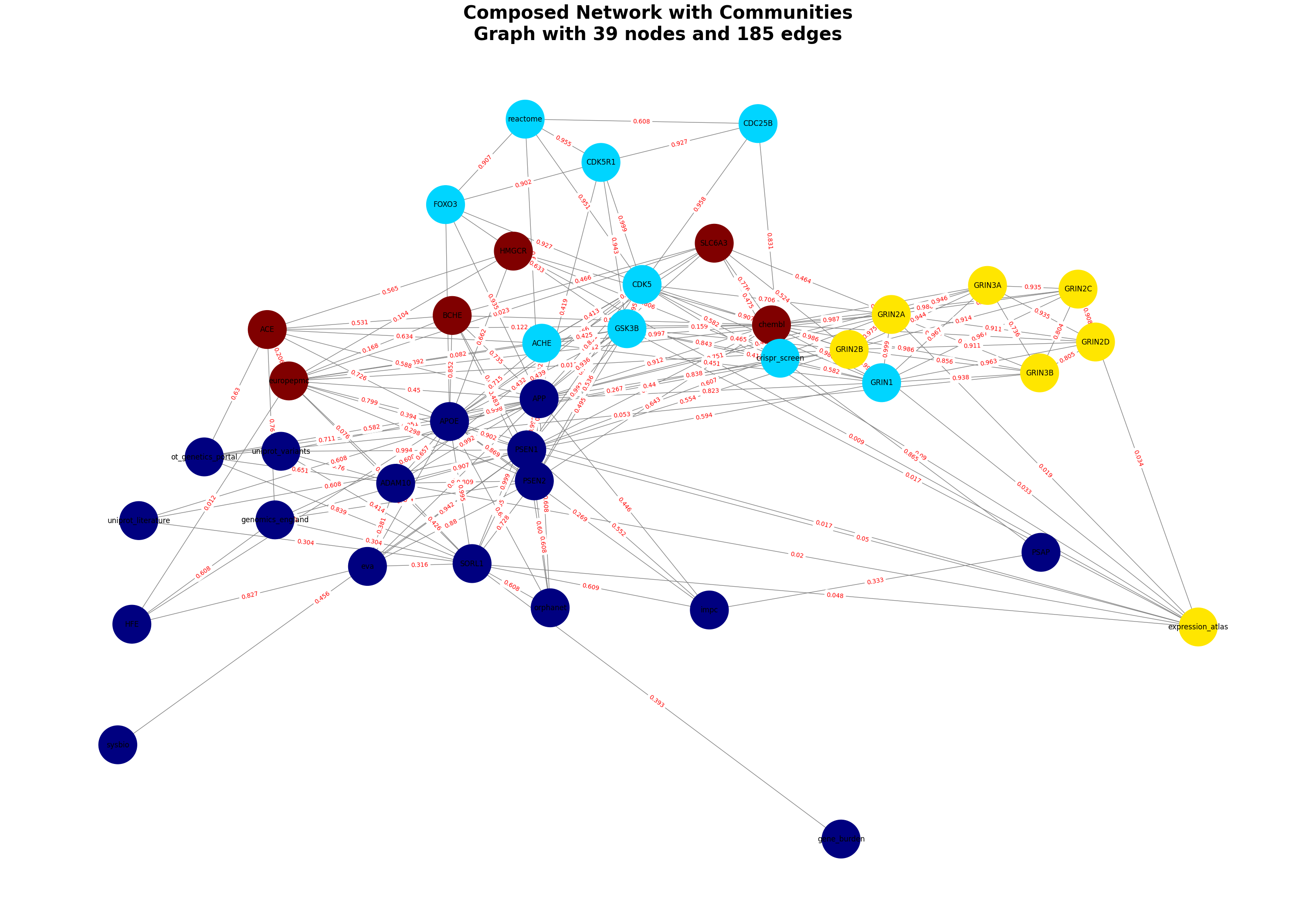
# Accessing the merged_df
merged_df2 = Alzheimer.get_merged_dataframe()
merged_df2.head()
| source | target | weight | type | |
|---|---|---|---|---|
| 0 | uniprot_variants | PSEN1 | 0.994 | disease-gene |
| 1 | uniprot_variants | APP | 0.951 | disease-gene |
| 2 | uniprot_variants | SORL1 | 0.414 | disease-gene |
| 3 | uniprot_variants | ADAM10 | 0.760 | disease-gene |
| 4 | PSEN1 | genomics_england | 0.955 | disease-gene |